

Journal of Creation 18(3):63–69, December 2004
Browse our latest digital issue Subscribe
Potentially decisive evidence against pseudogene ‘shared mistakes’
Summary
Recent discoveries of function in certain pseudogenes have led to the recognition, by some evolutionists, of widespread function in pseudogenes. The routine use of non-synonymous/synonymous ratios (KA/KS) does not lead to the indisputable conclusion that pseudogenes are simply pieces of junk DNA in a state of mutational ‘drift’. The complete unreliability of KA/KS as an indicator of pseudogene non-function is demonstrated by certain known functional pseudogenes.
The evolutionary storytelling of OR (olfactory receptor) genes progressively becoming pseudogenes as a consequence of the diminishing importance of olfaction in the course of primate evolution collapses in the face of recent research. Marmosets possess a strong sense of smell and numerous OR pseudogenes. Conversely, humans, for all their ‘degenerate’ sense of olfaction, have a set of ‘recently evolving’ OR genes.
Even if they are non-functional, orthologous primate urate oxidase (Uox) pseudogenes contain a phylogenetically-discordant, premature stop codon and a duplication. Pointedly, the striking discovery of the fact that the independently-derived guinea pig and human GULO pseudogenes have an astounding 36% identical ‘disablement’ is, if valid, as close as one can get to a resounding disproof of the entire evolutionistic ‘shared mistakes’ argument.
Pseudogenes are usually regarded as the disabled copies of protein-coding genes. For nearly twenty years, the evolutionist Edward Max1 has been highlighting pseudogenes as an insurmountably powerful argument for organic evolution and against special creation. This argument rests on the truth of the following three premises: 1) pseudogenes lack function, 2) while an Intelligent Designer may plausibly re-use the same designs for functional structures, it is unreasonable to suggest that an Intelligent Designer would create non-functional genes, let alone ones that share the same lesions from organism to organism, 3) owing to such ‘shared mistakes’, pseudogenes containing them could not have originated from independent inactivation events that occurred subsequent to the creation, but can only be explained through the common evolutionary ancestry of the organisms that bear them.
Almost everyone accepts the second premise. However, evidence has steadily been accumulating that undermines the first and third premises. For example, as discussed elsewhere,2 the non-functionality of pseudogenes, though presented as such, is not an established fact. There is a growing body of evidence to the contrary, as elaborated below. In addition, even if pseudogenes are in fact largely non-functional, lesions within them can originate independently.2 The present report extends and updates these considerations, devoting special attention to the primates’ olfactory receptor (OR) pseudogenes, the urate oxidase (Uox) pseudogenes and the GULO pseudogenes. Max1 has included all the foregoing as exemplary pseudogene-based evidences for organic evolution.
The irrelevance of non-synonymous/synonymous ratios (KA/KS)
This section introduces a methodology that is based entirely on the assumption of macroevolution in general and molecular evolution in particular. However, it can be shown that, even if this and auxiliary assumptions arc granted, the KA/Ks methodology is incapable of predicting the non-functionality of pseudogenes.
Evolutionists assume that all copies of genes (as well as their eventual pseudogene copies) originated from an ancestral gene. Therefore, they reason, the kinds of differences exhibited by corresponding genes and pseudogenes in different organisms (orthologs) are indicative of their prior evolutionary history. Known functional genes usually have very similar nucleotide sequences from one ortholog to another, and this is believed to be the outcome of natural selection maintaining the integrity of such sequences over long periods of time (purifying selection). In terms of specifics, those mutations that fail to change the peptide sequence itself (synonymous substitutions: abbreviated KS) are usually tolerated, but those that alter the peptide sequence (non-synonymous substitutions: abbreviated KJ are usually not (figure 1). Hence, according to standard evolutionary thinking, a low ratio of non-synonymous to synonymous substitutions (KA/K<<l) is the hallmark of a functional gene undergoing purifying selection. If, however, KA/KS >1, this is taken as an indicator that the functional gene is being subjected to active selection pressure and is probably in the process of evolving into another gene. Finally, KA/Ks =1 is taken to indicate a non-functional gene, or pseudogene, based on the supposition that pseudogenes are simply undergoing random mutational ‘decay’, exempt from the editing process of natural selection.
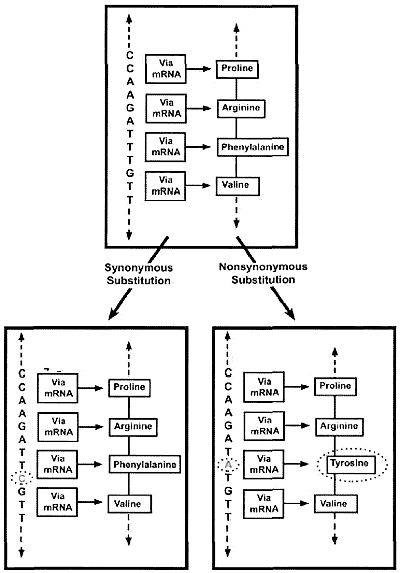
Pseudogene non-function not supported by (KA/KS)
The computation and interpretation of KA/KS is not straightforward. Estimates of KA/KS for the same gene or pseudogene can vary to a considerable extent depending on such things as the assumed nucleotide substitution rate, genomic context of the gene involved, etc.3 Recently, Zhang et al.4and Torrents et al.5 analyzed thousands of human pseudogenes. While inferred pseudogenes do tend to have KA/KS higher than those of genes, most of the values for pseudogenes are well under 1.0. Moreover, there is a significant zone of overlap of the KA/KS values of genes and pseudogenes, occurring at the range of KA/KS values between 0.1–0.5. It is acknowledged that, using this KA/KS methodology, ‘rapidly evolving’ functional genes may, ‘in a few cases’, be mistaken for pseudogenes. The presumed rarity of this situation6 is largely self-fulfilling, as it begs the question about the possibility of widespread pseudogene function. Pointedly, indisputably functional genes can, in whole or in part, have KA/KS at or near 1.0. Such is the case, for example, with the first exons of the human and baboon β-defensin genes.7
A recent review article,8 which carries a provocative title, draws startling conclusions about KA/KS relative to pseudogenes:
‘Pseudogenes exhibit evolutionary conservation of nucleotide sequence, reduced nucleotide variability, excess synonymous over non-synonymous nucleotide polymorphism, and other features that are expected in genes or DNA sequences that have functional roles.’9
The evidence concerning synonymous/non-synonymous substitutions is much stronger than is portrayed by these two authors. In fact, use of this technique is a holdover of the outdated ‘protein-encoding-only’ view of gene function. Pointedly, ratios of synonymous/non-synonymous substitution are largely, if not completely, inapplicable to some pseudogenes with known or potential functions. The Makorin1-p1 murine pseudogene10 performs a function completely different from that of its peptide-encoding paralogous (counterpart) gene, and it is very doubtful if any analysis of the KA/KS of the Makorin1-p1 pseudogene would cause the analyst to realize that this ‘severely crippled’ pseudogene is actually functional. Two snail pseudogenes are each functional in spite of being unable to code for even a full-length protein, or for any peptide at all,11 let alone for peptides that are, relative to those synthesized by their gene paralogs, conserved at synonymous sites! In addition, it has been shown that pseudogenes, despite being incapable of encoding peptides of appreciable length, can nevertheless encode very short peptide segments (8–11 amino acids length, with only a modest degree of sequence conservation over even this 8–11 amino acid span) that can at least potentially serve an immunobiological function.12 Clearly, the potential or actual function of a pseudogene can only be determined by direct experiment, and it is high time that evolutionists abandoned KA/KS as a ground for the a priori discounting of pseudogene function.
A broad range of pseudogene functions?
Disregarding pseudogenes for a moment, let us consider our state of knowledge of the functional genes in general. James D. Watson, writing on the 50th anniversary of his (and Francis Crick’s) discovery of the helical structure of the DNA molecule, commented as follows:
‘The most humbling aspect of the Human Genome Project so far has been the realization that we know remarkably little about what the vast majority of human genes do.’13
Now if the foregoing is true of genes, how much more so of pseudogenes! At very least, pseudogenes have not been fairly and objectively analyzed:
‘An extensive and fast-increasing literature does not justify a sharp division between genes and pseudogenes that would place pseudogenes in the class of genomic “junk” DNA that lacks function and is not subject to natural selection.’14
In support of this position, Balakirev and Ayala15 present several lines of evidence pointing to pseudogene function. These include the blurred gene/pseudogene boundary in organisms (notably Drosophila), the widespread transcription of pseudogenes, the pseudogene-based enhancement of immunoglobin gene diversity, and the regulatory role of genes performed by some pseudogenes. Pseudogenes can serve non-coding functions as CpG islands (stretches of DNA that are enriched in cytosine and guanine at the expense of adenine and thymine), enhancers (a type of gene regulatory sequence), and matrix attachment regions (a DNA structural feature).
These two iconoclastic authors also warn against the standard assumption that pseudogenic features (variously described as ‘lesions’, ‘disablements’ and ‘mistakes’) are necessarily incompatible with pseudogene function:
‘How pervasive are “functional” pseudogenes? Many pseudogenes have been identified in all sorts of organisms on the grounds that they are duplicated genes that exhibit stop codons or other disabling mutations in their DNA sequences, so that they cannot have the full function of the original genes from which derived. In many of these cases, however, it remains unknown, because it has not been investigated, whether the pseudogenes, described only on the basis of DNA sequences, may have acquired regulatory or other functions, or play a role in generating genetic variability’ [emphasis added].14
However, Balkirev and Ayala do not go far enough in their attempt to come to grips with the limited significance of ‘lesions’ relative to pseudogene function. As demonstrated elsewhere16—and semantics aside—there is actually a whole set of indisputably functional genes that qualify as functional pseudogenes in that they have major pseudogenic features such as premature stop codons, indels (insertions and deletions), etc., that are circumvented by genomic recoding processes (e.g. stop codon readthrough, frameshifting, etc.). Moreover, a variety of recent developments in our understanding of the functions of junk DNA in general, elaborated elsewhere,17 have implications for the potential widespread functionality of pseudogenes. These include the previously unsuspected genomic ‘parallel universe’ of RNA-only functions as well as the unexpectedly common antisense transcription of human DNA.
Based on Balkirev and Ayala’s most recently quoted statements, it is plain to see that pseudogenes generally appear to lack function primarily because very few of them have been carefully studied for function. It is no more complicated than that! In fact, these two researchers are willing to go as far as recognizing a generalized functionality of pseudogenes:
‘There seems to be the case that some functionality has been discovered in all cases, or nearly all, whenever this possibility has been pursued with suitable investigations. One may well conclude that most pseudogenes retain or acquire some functionality and, thus, that it may not be appropriate to define pseudogenes as non-functional sequences of genomic DNA originally derived from functional genes, or as “genes that are no longer expressed but bear sequence similarity to active genes (99, p. 114) [sic] [emphasis added]”.’14
With the realization of the fact that pseudogenes can actually be unconventionally behaving genes, the following consideration takes on further significance:
‘Pseudogenes may lose some specific functions but retain others, and even acquire new ones, which may not be simply recognizable.’14
Primate olfactory receptor (OR) pseudogenes vs degree of olfaction
Compared to many primates, humans have a diminished sense of smell and a large number of OR pseudogenes. This has led to the notion that, as olfaction supposedly became less and less important during the course of primate evolution, the relative number of OR pseudogenes has increased. This follows from the supposition that, in a primate largely reliant on a strong sense of smell for survival, an inactivated OR gene (OR pseudogene) would likely be disadvantageous and its bearer removed by natural selection. In a primate whose sense of smell is less important for its survival, the ‘pseudogenization’ of OR genes would be more commonly tolerated by the process of natural selection. Consequently, over time, OR pseudogenes would tend to accumulate in the genome of the latter. It makes for a nice evolutionary story, and has been repeated by Max.1
A recent study18 overturns the notion that there is any kind of straightforward relationship between the importance of olfaction in a primate and the extent of inactivation, or ‘pseudogenization’, of its OR genes:
‘It has generally been assumed that OR pseudogene formation has a close relationship to olfactory function. However, it is likely that there is a background rate of OR gene turnover (duplications and pseudogene formation) in all lineages, and that for many of these events the functional consequences are minimal … These results provide a contrast to previous studies, and show that in spite of the keen olfactory sense of marmosets, they harbour many OR pseudogenes.’19
To further complicate matters, an evolutionary study comparing mouse, chimp, and human OR genes indicates that certain human OR genes are experiencing inferred positive selection.20 This admittedly surprising finding has prompted ad hoc suggestions that certain OR genes may double as agents involved in sexual selection, dietary changes, etc.21 In any case, the belief that ‘pseudogenization’ of OR genes correlates with partial loss of olfaction is clearly shown to be a rather hasty evolutionistic generalization.
The urate oxidase (Uox) pseudogenes: ‘shared mistakes’ without common ancestry
The absence of urate oxidase causes an increase in the levels of uric acid in the body. It has been suggested that this increased level is actually beneficial in the protection of the body against oxidative stress etc. Whether or not such speculations have any merit within the creationist–diluvialist paradigm is not clear at present.
The paralogous Uox pseudogenes, believed to be the remnants of a gene that once coded for urate oxidase, share a number of identical ‘disablements’. A recent study22 goes far beyond the earlier studies of the Uox pseudogenes that had been cited and discussed by Max.1 The new evidence does not, for the most part, support his old evidence. A series of ‘lesions’ are specific to given primates. Moreover, some of them violate the evolutionary nested hierarchy by being absent in the primitive phylogenetic state and present in an intermediate phylogenetic state, only to be absent in a derived state.
Perhaps the most prominent ‘shared mistake’ among pseudogenes overall is the premature stop codon. Two (of the six23) premature stop codons now known for the Uox pseudogenes (at codons 33 and 187) do follow an evolutionary ‘shared mistakes’ deployment. Members of the orang-gorilla-chimp-human clade share the codon 33 premature stop codon, while the one at codon 187 is found in gorillas, chimps and humans. In contrast, three others (at codons 18, 167 and 197) are unique to one type of primate only, and, based on post hoc evolutionary reasoning, are imagined to have originated after each respective primate had branched off its ancestral lineage. Finally, one of the newly discovered premature stop codons in the Uox pseudogene has an evolutionarily impossible deployment (figure 2), described as follows:
‘The nonsense mutation (TGA) at codon 107 is, however, more complicated than others. It occurs in the gorilla, the orangutan, and the gibbon, and therefore requires multiple origins of this nonsense mutation.’24
This is not an isolated occurrence. The human and sheep orthologous P2 pseudogenes share a coincidental premature stop codon without the possibility of common evolutionary ancestry.25 It is not difficult to understand the independent formation of coincidental premature stop codons (with or without the ‘fact’of evolution) once one realizes the limited number of possibilities for their origination. There are only three possible stop codon triplets, and they occur at subequal frequencies (with TGA the most common). Only a few of the 20 amino acid codons can readily mutate to a premature stop codon.26 The recent analysis of the Uox pseudogenes reinforces this conclusion. The CGA codon (arginine) mutated to the premature stop codon TGA,27 and this was no doubt facilitated by the well-known propensity of CG doublets to mutate. Of course, had this particular premature stop codon occurred in a nested hierarchy, it would have been cited as an evolutionary ‘shared mistake’, and the CG issue would not have been raised. To be consistent, evolutionists should discount all ‘shared mistakes’ that have inferred CG precursors, not just those that contradict evolutionary schemes. Needless to say, this is not done. Of course, the known propensity for certain DNA sequences to mutate goes far beyond the CG doublet, and this is elaborated later in this paper.

Figure 2. Evolutionary nested hierarchy, expressed as a Venn diagram,
with the chimp-gorilla clade as the crown group (according to data from the Uox
gene in baboon and the orthologous pseudogenes found in more ‘derived’
primates). Two major ‘shared mistakes’ of the Uox pseudogene
[the premature stop codon at codon 107, and the GGGATGCC duplication (D) at nucleotides
911–917 of intron 4] violate this nested hierarchy, eliminating the possibility
of their origins from a common ancestor. Note that the respective primitive character
states [absence of the premature stop codon, symbolized by (Go), and the no duplication
(ND) condition] reappear in the most ‘derived’ primates (smallest ovals).
|
A phylogenetically discordant duplication (shown as D in figure 2) has its own blow to strike against the whole evolutionary ‘shared mistakes’ argument:
‘One exceptional change is a duplicated segment of GGGATGCC in intron 4, which is shared by the gorilla and the orangutan. However, because this change is phylogenetically incompatible with any of the three possible sister-relationships among the closely related trio of the human, the chimpanzee, and the gorilla, it might result from two independent duplications. Alternative, though less likely, a single duplication occurred in the ancestral species of the great apes and had been polymorphic for a sufficiently long time to permit fixation of the duplicated form in the orangutan and the gorilla on the one hand and loss in the human and chimpanzee on the other hand.’28
Moreover, in contradiction to the widely (but not universally) accepted view that humans are the sister group of chimps, the upstart Uox pseudogenes support a close relationship between the chimp and the gorilla:
‘It is to be noted that the sister-relationship between the chimpanzee and the gorilla to the exclusion of the human is supported by 100% bootstrap probability. In fact, there are six phylogenetically informative sites supporting the chimpanzee-gorilla clade but none supporting the alternatives among the human-chimp-gorilla relationships.’28
In view of the contradictory scenarios involving human-chimp-gorilla relationships, it is evident that pseudogene ‘shared mistakes’ are providing conflicting phylogenetic information.
Introduction to vitamin C biosynthesis
In order to understand yet another pseudogene, one that is presumably related to the inability of humans to produce their own vitamin C, a brief introduction is presented. Ascorbic acid (vitamin C) is an essential micronutrient that performs a variety of functions in the body. Humans, simian primates, guinea pigs and a few other vertebrates are incapable of synthesizing their own vitamin C, and so require dietary sources of this vitamin. The conventional recommended intake of vitamin C in humans is an order of magnitude below the amount synthesized by those mammals (the vast majority) capable of producing it,29 and this has prompted controversial suggestions (e.g. Linus Pauling) that humans should take vitamin C at daily gram-level doses.
Nishikimi and Yagi30 have summarized our current knowledge of ascorbic acid biosynthesis among vertebrates. The terminal step involves the conversion of L-gulono-γ-lactone to L-ascorbic acid through the action of the enzyme L-gulono-γ-lactone oxidase (GLO or GULO). GULO has been found in certain fish, amphibians, reptiles, birds, egg-laying mammals, and most kinds of eutherian mammals. It has long been known that, unlike the true simians, prosimians (tree shrew, galago, slow loris and pottos) are able to produce GULO.31 It is notably absent in humans, apes, monkeys and guinea pigs. Scurvy, which occurs only when dietary sources of ascorbic acid are insufficient to compensate for the body’s inability to synthesize it, is depicted as an unusual type of inborn error of metabolism.
The GULO pseudogenes: numerous ‘shared mistakes’ without common ancestry
Many of those mammals found unable to synthesize ascorbic acid have regions of their genome that are believed to correspond to parts of the functional GULO gene that is found in those mammals found capable to synthesizing GULO, and thus vitamin C. Evolutionists have cited these apparently vestigial remnants of GULO to make dysteleological arguments against an Intelligent Designer. In addition, they1 have argued that lesions found in common between the orthologous GULO pseudogenes of simian primates (‘shared mistakes’) argue strongly for their origins from a common ancestor, and all but rule out an independent inactivation of the GULO gene among different simian primates (including humans).
Previous studies of the orthologous primate GULO gene and pseudogene have focused on those parts of a few exons that appear to correspond between humans and rats. A more recent study32 is much more comprehensive. It is now believed that, relative to the 12 exons that comprise the functional rat GULO gene, the human GULO pseudogene is limited to counterparts of exons 4, 7, 9, 10, and 12. Owing to the fact that the guinea pig and the simian primates are obviously not sister groups (fig. 3), it is impossible for the guinea pig GULO pseudogene and the human GULO pseudogene to have both originated from the same ancestral pseudogene. Furthermore, not only are the inactivations of GULO in the guinea pig and primates clearly independent events based on phylogenetic analysis (fig. 3), but also on inferred evolutionistically believed times of inactivation. Summarizing earlier studies, Nishikimi and Yagi,33 using ‘molecular clocks’, estimate that the guinea pig lost GULO function less than 20 million years ago. In contrast, the separate inactivation of the GULO gene in primates allegedly occurred between the time of simian-prosimian divergence (50–65 million years ago) but before the Old/New world monkey divergence (35–45 million years ago).
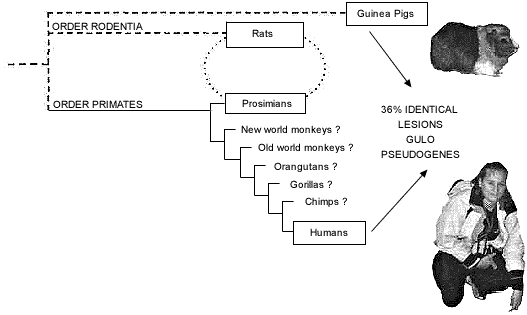
In spite of all this, a recent comparison of the independently derived guinea pig GULO pseudogene and the human GULO pseudogene has produced the following staggering discovery:
‘When the human and guinea pig sequences (647 nucleotides in total) of the regions of exons 4, 7, 9, 10, and 12 were compared, we found 129 and 96 substitutions in humans and guinea pigs, respectively, when compared with the rat sequences (Fig. 2) [sic] [in original article]. The same substitutions from rats to both humans and guinea pigs occurred at 47 nucleotide positions among the 129 positions where substitutions occurred in the human sequences’ [emphasis added].34
Detailed examination of the relevant sequence35 (47 positions among the 647 nucleotides) reveals no obvious pattern suggestive of a straightforward explanation for this abundance of parallel nucleotide substitutions. The distribution of the 47 positions is: Exon 4 (6 positions), Exon 7 (10), Exon 9 (10), Exon 10 (9) and Exon 12 (12). The positions themselves are spread out subequally across each exon. None of the 47 positions occur more than two in a row, and there are only two sites (four total positions) in which the positions occur two in a row. There are only four indels, none more than 3 nucleotides long, in the five exons of the three collective sequences. None of the 47 positions is associated with an indel. In fact, none of the 47 positions occur within three nucleotides of an indel. All sixteen possible doublets of nucleotides are associated with the 47 positions of parallel mutation, and there is no strongly preferred doublet tending to mutate to any one of the 47 positions. Furthermore, only 3 of the 47 positions are associated with the highly mutable CG doublet. None of the 47 positions are associated with homopolymeric runs (e.g. AAAAAA …). Four same-site stop codons (3 TGA and 1 TAA) have been independently created in the guinea pig and human GULO pseudogenes, if all three overlapping ORFs (open reading frames) are considered. None of these four stop codons could have originated from a CG doublet.
Let us now take the pseudogene ‘shared mistake’ argument to its logical conclusion. The unexpected degree of identicalness between the ‘lesions’ of the guinea pig GULO pseudogene and those of its counterpart in the higher primates (including humans) leads to the preposterous conclusion that humans are more closely related to guinea pigs than to prosimian primates!
GULO pseudogenes’ ‘shared mistakes’ from mutational hotspots
Even if one accepts organic evolution, one must concede that the astounding degree of identicalness of the ‘mistakes’ common to the guinea pig and human GULO pseudogenes could not possibly have resulted from evolutionary ancestry. If the Inai et al.32 analysis is valid, and there is no reason for questioning its validity, it falsifies Max’s1 long-promoted pseudogene ‘shared mistakes’ argument. If a strong pattern of pseudogenic ‘shared mistakes’ can happen even once in an evolutionarily impossible manner, it can also happen again and again in an evolutionarily consistent manner. Now, more than ever, Occam’s razor dictates that ‘shared mistakes’ be approached in terms of parallel mutations rather than common evolutionary ancestry.
Of course, it is virtually inconceivable that these many identical nucleotide substitutions have arisen solely by chance:
‘Assuming an equal chance of substitution throughout the sequences, the probability of the same substitutions in both humans and guinea pigs occurring at the observed number of positions and more was calculated to be 1.84 X 10-12. This extremely small probability indicates the presence of many mutational hot spots in the sequences.’35
It has long been known that mutations are quite non-random in occurrence, but the variety and complexity of mutational hotspots has seldom been appreciated. Rogozin et al.36 have recently summarized our current knowledge of experimentally induced mutations. Many nucleotide motifs other than the earlier-discussed CG doublet can serve as mutational hotspots. It is now known that the sequence content tens of bases away from a given motif can influence the degree of its hotspot behaviour. Moreover, the propensity of nucleotide motifs to be mutational hotspots varies from gene to gene and from one region of the genome to another. Moreover, the foregoing considerations do not even touch the higher-level features of gene or chromatin structure as causes of mutational hotspot behaviour.37 The large relative number of parallel mutations in the guinea pig and primate GULO pseudogenes cannot be said to be unprecedented. Experimental evidence has already demonstrated that nucleotide substitutions (as well as indels, for that matter) can, unexpectedly, occur in a very strongly concerted manner.38
Conclusions
It is obvious that the major premises on which evolutionary pseudogene-based arguments rest are steadily crumbling. Some evolutionists are now recognizing the widespread functionality of pseudogenes. In the light of this fact, the notion of ‘shared mistakes’ yields to ‘shared engineering and/or artistic similarities’—as is recognized by creationist scientists for all homologies encountered between living organisms. Pseudogenes must be recognized as non-canonical genes as well as truly disabled genes. The two categories are not mutually exclusive, and the creationist scientist must accommodate both eventualities.
The striking degree of identicalness between the ‘lesions’ of presumably non-functional pseudogenes, unrelated by evolutionary ancestry, clearly dispenses with organic evolution as a necessary explanation for this overall phenomenon. Moreover, it reopens the consideration of such pseudogenes being one-time functional genes that became independently disabled sometime after the Fall. Much more must be learned about the thousands of pseudogenes in various genomes before detailed generalizations about them can be made in a scientific creationist context.
References
- Max, E.E., Plagiarized errors and molecular genetics, 5 May 2003.
- Woodmorappe, J., Are pseudogenes ‘shared mistakes’ between primate genomes? Journal of Creation14(3):55–71, 2000.
- Ina, Y., New methods for estimating the numbers of synonymous and non-synonymous substitutions, J. Molecular Evolution 40:190–226, 1995.
- Zhang, A., Harrison, P.M., Liu, Y. and Gerstein, M., Millions of years of evolution preserved: a comprehensive catalog of the processed pseudogenes in the human genome, Genome Research 13:2541–2558, 2003.
- Torrents, D., Suyama, M., Zdobnov, E. and Bork, P., A genome-wide survey of human pseudogenes, Genome Research 13:2559–2567, 2003.
- Torrents et al., ref. 5, p. 2564.
- Semple, C.A.M., Rolfe, M. and Dorin, J.R., Duplication and selection in the evolution of primate β-defensin genes, Genome Biology 4(5)R31.1–R31.11, 2003.
- Balakirev, E.S. and Ayala, F.J., Pseudogenes: are they ‘junk’ or functional DNA? Annual Review of Genetics 37:123–151, 2003. The very title of this article would have, only a few years ago, been almost on a par with the following: ‘The Earth: is it spherical or flat?’
- Balakirev and Ayala, ref. 8, p. 123.
- Woodmorappe, J., Pseudogene function: regulation of gene expression, Journal of Creation 17(l):47–52, 2003.
- Woodmorappe, J., Pseudogene function: more evidence, Journal of Creation 17(2):15–18, 2003.
- Woodmorappe. J., The potential immunological functions of pseudogenes and other junk DNA, Journal of Creation 17(3):102–108, 2003.
- Watson, J.D., DNA: The Secret of Life, Alfred A. Knopf, New York, p. 217, 2003.
- Balakirev and Ayala, ref. 8, pp. 137–138.
- Balakirev and Ayala. ref. 8, pp. 123–151.
- Woodmorappe, J., Unconventional gene behavior and its relationship to pseudogenes, Proceedings of the Fifth International Conference on Creationism, Technical Volume, pp. 505–514, 2003.
- Woodmorappe, J., Junk DNA indicted, Journal of Creation 18(1):27–33, 2004.
- Whinnett, A. and Mundy, N.I., Isolation of novel olfactory receptor genes in marmosets (Callithrix): insights into pseudogene formation and evidence for functional degeneracy in non-human primates, Gene 304:87–96, 2003.
- Whinnett and Mundy, ref. 18, pp. 95, 87.
- Clark, A.G., Glanowski, S., Nielsen, R. et al., Inferred nonneutral evolution from human-chimp-mouse orthologous gene trios, Science 302:1960–1963,2003.
- Pennisi, E., Genome comparisons hold clues to human evolution, Science 302:1867–1877, 2003.
- Oda, M., Satta, Y., Takenaka, O. and Takahata, N., Loss of urate oxidase activity in hominoids and its evolutionary implications, Molecular Biology and Evolution 19(5):640–653, 2002.
- Oda et al., ref. 22, p. 650.
- Oda et al., ref. 22, p. 643.
- Woodmorappe, ref. 2, p. 70. See the discussion surrounding reference 97 therein.
- Woodmorappe, ref. 2, p. 61.
- Oda et al. ref. 22, p. 645–647.
- Oda et al., ref. 22, p. 642.
- Challem, J.J. and Taylor, E.W., Retroviruses, ascorbate, and mutations, in the evolution of Homo Sapiens, Free Radical Biology & Medicine 25(1):131, 1998.
- Nishikimi, M. and Yagi, K., Biochemistry and molecular biology of ascorbic acid biosynthesis, Subcellular Biochemistry 25:17–39, 1996. For a sketch of the complete biochemical pathway that culminates in the synthesis of Vitamin C, see figure 1 (p. 19) therein.
- Nakajima, Y., Shantha, T.R. and Bourne, G.H., Histochemical detection of L-gulonolactone, Histochemie 18:293–301, 1969; see especially p. 300.
- Inai, Y., Ohta, Y. and Nishikimi, M., The whole structure of the human non-functional L-gulono-γ-lactone oxidase gene—the gene responsible for scurvy—and the evolution of repetitive sequences thereon, J. Nutritional Science and Vitaminology (Tokyo) 49(5):315–319, 2003.
- Nishikimi and Yagi, ref. 30, p. 34.
- Inai et al., ref. 32, p. 316.
- Inai et al., ref. 32, p. 317.
- Rogozin, I.B.. Babenko, V.N., Milanesi. L. and Pavlov, Y.I., Computational analysis of mutation spectra, Briefings in Bioinformatics 4(3):210–227, 2003.
- Rogozin et al., ref. 36, p. 221.
- Woodmorappe, ref. 2, p. 65. See also refs. 144–146 cited and discussed therein.
Readers’ comments
Comments are automatically closed 14 days after publication.