

Journal of Creation 13(1):5–7, April 1999
Browse our latest digital issue Subscribe
Decoding and editing design: double-sieve enzymes
All living organisms contain literally encyclopedic quantities of complex, specific information. To store this information, living things have by far the most compact information storage/retrieval system known: the nucleic acid/protein system. The master blueprint or recipe is coded on enormous molecules of DNA (deoxyribonucleic acid).1 A codon, or sequence of three of the four types of DNA ‘letters’ (nucleotides), codes for one of the 20 types of protein ‘letters’ (amino acids). A gene is defined as a sequence of nucleotides coding for a single protein, or a subunit of a multicomponent protein. Even the smallest known genome of any free-living organism, Mycoplasma genitalium, contains 482 genes comprising 580,000 nucleotides. 2
The decoding (translation) requires many components, including complex editing machinery to correct errors. But the famous philosopher of science, Sir Karl Popper (1902–1994), pointed out:
‘…the machinery by which the cell (at least the non-primitive cell, which is the only one we know) translates the code consists of at least fifty macromolecular components which are themselves coded in the DNA. Thus the code can not be translated except by using certain products of its translation. This constitutes a baffling circle; a really vicious circle, it seems, for any attempt to form a model or theory of the genesis of the genetic code.’3
The obvious conclusion is that the decoding must have been functional from the beginning, otherwise life could not exist.
Decoding molecules

One of the many types of molecules needed are the transfer ribonucleic acid (tRNA) molecules. These are the molecules which link the right amino acid with the right codon. They comprise about 80 nucleotide ‘letters’, three of which are called the anticodon. The anticodon links to the corresponding codon on the messenger RNA (mRNA), which in turn has relayed the correct code from the DNA. Thus the tRNAs can transfer the right amino acids to the right place in the growing peptide chain, as coded in the mRNA.4 Also, the amino acid is bonded to the tRNA in such a way as to be activated, i.e. to have a high chemical potential—this is necessary so it will form a peptide bond to the adjacent amino acid in the polypeptide. Free amino acids have almost no tendency to form polypeptides by themselves; rather, the tendency is for the reverse to happen.5
There are also enormous chemical hurdles for any evolutionary explanation of the origin of nucleic acids from a hypothetical primordial soup.6, 7 And even if we granted that RNA could form spontaneously, there is a huge hurdle in linking the right amino acid to the right anticodon by naturalistic means. If the links are not correct, the entire decoding machinery would decode the wrong message, or no message at all, meaning that the organism could not manufacture vital enzymes. However, there is no chemical reason for any particular anticodon to link to any particular amino acid. In fact, they are at the opposite ends of the tRNAs, precluding any chemical interaction. Again, they must have been fully functional from the beginning.
Synthesizing the tRNAs
Living organisms do not, and could not, rely on random chemistry to synthesize the tRNAs. Rather, the right amino acid is activated and linked in two steps to the right tRNA by aminoacyl-tRNA synthetases (aaRSs).8 First, chemical energy is supplied by adenosine triphosphate (ATP), which was formed elsewhere by ATP synthase, an enzyme containing a miniature rotary motor, F1-ATPase. 9, 10, 11, 12 ATP reacts with the amino acid to form a mixed carboxylic-phosphoric anhydride.13 Secondly, the aminoacyl group forms an ester with the 3’-hydroxyl of the ribose in the terminal adenosine of the tRNA.8,13
Editing—double sieve enzymes
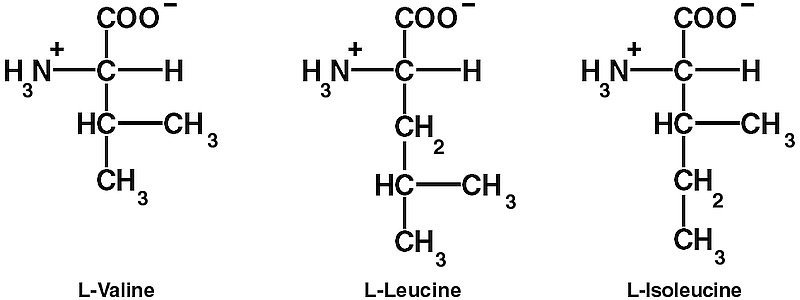
However, these steps are not enough to ensure the required high decoding fidelity (error rates of 1/2400 to 1/40,000). The aaRSs also edit the final products to make sure that the right amino acid is linked to the right tRNA. One difficulty is discriminating between chemically similar amino acids. In particular, L-valine (Val) and L-isoleucine (Ile) differ by only one methylene (CH2) group. Double Nobel laureate Linus Pauling (1901–1994), calculated that since the CH2 group has a hydrophobic binding energy of only about 4 kJ/mol, the error rate for replacing Ile with Val would be about one in five.14 So it is thermodynamically impossible for ordinary one-step recognition to achieve the error rate of 1/3,000 observed in isoleucyl-tRNA synthetase (IleRS).15, 16, 17, 18
However, an error substituting Ile for Val can be biologically harmful or even catastrophic. Even a single Ile–Val mutation in the core of ribonuclease T1 reduces its stability because of ‘a loss of favorable packing interactions of the side chain in the folded form of the protein.’19 Such a mutation in the hydrophobic core of chymotrypsin inhibitor 2 changes the free energy of unfolding (DDGU–F) by 5.0 ± 0.4 kJ/mol on average.20 And a single Ile–Val mutation in the interior of human lysozyme results in less resistance to denaturation ((DDG from -1.5 – -5.0 kJ/mol).21 This mutation also increases susceptibility to lung cancer22 and affects Human Immunodeficiency Virus–1 drug resistance.23
Another problem cited by Pauling is that while an enzyme’s binding site can easily exclude molecules that are larger by steric hindrance, how can it exclude molecules that are smaller?14,15
Alan Fersht first proposed a solution in 1977: a ‘double-sieve’ editing mechanism.24 A coarse sieve would exclude larger amino acids from being activated, but allow the right amino acid and the smaller ones to be activated. Then a fine sieve would hydrolyse the products of the smaller amino acids (see diagram below).

In 1998, Nureki et al. demonstrated this double-sieve mechanism in IleRS. They used X-ray diffraction (XRD) techniques to solve the crystal structure of Thermus thermophilus IleRS, as well as its complexes with Ile and Val. IleRS is a huge L-shaped molecule measuring about 100 Å x 80 Å x 45 Å , and belongs to the space group C2.8
IleRS contains a characteristic nucleotide binding fold, the Rossmann fold, in the centre. The ‘coarse sieve’ is a cleft in the Rossmann fold with two characteristic four-amino-acid sequences that bind ATP. The cleft also binds L-Ile at the bottom — its hydrocarbon groups and the NH3+ and COO– groups are recognized by strategically placed amino acid residues of the enzyme. This site is able to exclude larger amino acids by steric hindrance, including L-leucine, although this differs from Ile only in the placement of the methyl group on the side chain. This contrasts with ordinary laboratory organic chemistry, where ‘Leucine and isoleucine are particularly difficult to separate.’ 25
The fine sieve is another part of the Rossmann fold, the Ins-2 structural domain, which contains another deep cleft. XRD detected Val in this cleft in the L-valine-IleRS complex, but never any Ile in the L-isoleucine-IleRS complex — the cleft is simply too small. The incorrect Val products are hydrolysed here, but the correct Ile products are protected.
Nureki et al. demonstrated this by constructing a mutant IleRS which lacked 47 amino acid residues including a tryptophan (Trp232) of the L-valine-specific pocket.8 This completely destroyed the editing ability. In another experiment, Nureki et al. mutated just two amino acids (replacing Thr243 and Asn250 with alanine) of E. coli IleRS, which again completely destroyed the editing ability. Previous work had shown that even a single mutation (replacing Tyr403 with Phe) greatly reduces the editing ability of E. coli IleRS.26
Other aaRSs also have editing activity, including ValRS, which deacylates errant threonine products.27
Evolutionary bias
Unfortunately, the brilliant paper of Nureki et al.8 was spoiled when the authors went with the common secular flow, and genuflected to the idol of today—the Unholy Trinity of Time, Chance and Natural Selection. They wrote:
‘ … it is interesting from an evolutionary viewpoint that all of the enzymes catalyzing the central steps of Ile-Val biosynthesis and metabolism do not distinguish, or can neglect the difference, between the two aliphatic amino acids, as was observed for the first catalytic site of IleRS. This finding implies that a putative ancestral enzyme of IleRS and ValRS might have actually had a similar dual specificity for L-isoleucine and L-valine in a primordial genetic code system.’ 28
Of course, a good designer will often use similar machinery to make similar products, 29 and it makes sense especially with the extremely close chemical similarity of Ile and Val.25 And their statement is merely ‘just-so’ story telling, lacking even the slightest evidence. It is no substitute for explaining exactly how such an editing site could evolve by natural selection. This site requires many amino acids in precise sequences before it could work at all, thus exhibiting a hallmark of design—what biochemist Michael Behe, in his book Darwin’s Black Box, termed irreducible complexity.30 The problem is especially acute in this case—since natural selection equals differential reproduction, if there is poor editing, then accurate reproduction of successful traits is impossible. Error catastrophe is more likely.29, 31, 32
References
- For an instructive illustration, see Gitt, W., Dazzling Design in Miniature , Creation 20(1):6, 1997. Return to text.
- Fraser, C.M., et al. The minimal gene complement of Mycoplasma genitalium’, Science 270(5235):397–403, 1995; Perspective by A. Goffeau, Life with 482 genes, same issue, pp. 445–6. Return to text.
- Popper, K.R., Scientific reduction and the essential incompleteness of all science; in Ayala, F. and Dobzhansky, T., eds., Studies in the Philosophy of Biology, University of California Press, Berkeley, p. 270, 1974. Return to text.
- For a good description, see Denton, M., Evolution: a Theory in Crisis, Adler & Adler, Bethesda, Maryland, ch. 10, 1985. Return to text.
- Sarfati, J.D., Origin of life: the polymerization problem. CEN Tech. J. 12(3):281–284, 1998. Return to text.
- Mills, G.C. and Kenyon, D.H., The RNA World: A Critique, Origins and Design 17(1): 9–16, 1996. Return to text.
- Sarfati, J.D., Self-Replicating Enzymes? Journal of Creation. 11(1):4–6. Return to text.
- Nureki, O. and nine others, Enzyme structure with two catalytic sites for double-sieve selection of substrate, Science 280(5363):578–582, 1998. Return to text.
- Hiroyuki Noji et al., Direct observation of the rotation of F1-ATPase. Nature 386(6622):299–302, 1997; perspective by Block, S., Real engines of creation, same issue, pp. 217–219. Return to text.
- Boyer, P., The binding change mechanism for ATP synthesis—some probabilities and possibilities, Biochim. Biophys. Acta 1140:215–250, 1993. Return to text.
- Abrahams, J.P. et al., Structure at 2.8 Å … resolution of F1-ATPase from bovine heart mitochondria. Nature 370(6491):621–628, 1994. Comment by Cross, R.L. Our primary source of ATP. Same issue, pp. 594–595. Return to text.
- Sarfati, J.D., Design in living organisms: Motors , Journal of Creation. 12(1):3–5, 1998. Return to text.
- Karlson, P., (tr. Doering, C.H.), Introduction to modern biochemistry, 4th ed., Academic Press, NY, London, pp. 145–146, 113, 1975. Return to text.
- Pauling, L., in Festschrift Arthur Stoll, Birkhäuser Verlag, Basel, Switzerland, p. 597, 1958; cited in Nureki et al., Ref. 8. Return to text.
- Fersht, A.R., Sieves in sequence, Science 280(5363):541 (comment on Nureki et al., Ref. 8), 1998. Return to text.
- Freist, W., Pardowitz, I. and Cramer, F., Isoleucyl-tRNA synthetase from baker’s yeast: multistep proofreading in discrimination between isoleucine and valine with modulated accuracy, a scheme for molecular recognition by energy dissipation, Biochemistry 24(24):7014–7023, 1995. Return to text.
- Loftfield, R.B., Biochem. J. 89:82–92, 1963; cited in Freist et al., Ref. 16. Return to text.
- Loftfield, R.B. and Vanderjagt, D., Biochem. J. 128:1353–1356, 1972; cited in Freist et al., Ref. 16. Return to text.
- Sneddon, S.F. and Tobias, D.J., 1992. The role of packing interactions in stabilizing folded proteins. Biochemistry 31(10):2842–2846. Return to text.
- Jackson, S.E. et al., 1993. Effect of cavity-creating mutations in the hydrophobic core of chymotrypsin inhibitor 2. Biochemistry 32(43):11259–11269. Return to text.
- Takano, K. et al., 1995. Contribution of hydrophobic residues to the stability of human lysozyme: caolimetric studies and x-ray structural analysis of the five isoleucine to valine mutants. J. Mol. Biol. 254:62–76. Return to text.
- Zhang, Z.Y. et al., 1996. Cancer Res. 56:3926; cited in Nureki et al., Ref. 8. Return to text.
- Farrash, M.A. et al., 1994. J. Virol. 68:233; cited in Nureki et al., Ref. 8. Return to text.
- Fersht, A.R., 1977. Enzyme Structure and Mechanism, Freeman, San Francisco, p. 283; cited in Fersht, Ref. 15. Return to text.
- Karlson, Ref. 13, p. 27. Return to text.
- Schmidt, E. and Schimmel, P., 1995. Residues in a Class I tRNA synthetase which determine selectivity of amino acid recognition in the context of tRNA. Biochemistry 34(35):11204–11210. Return to text.
- Eldred, E.W. and Schimmel, P., 1972. J. Biol. Chem. 247:2961; cited in Nureki et al., Ref. 8. Return to text.
- Nureki et al., Ref. 8, p. 581. [See p. 17 this journal.] Return to text.
- ReMine, W.J., The Biotic Message, St. Paul Science, St. Paul, MN, passim, 1993; see review by Batten, D., Journal of Creation. 11(3):292–298, 1997. Return to text.
- Behe, M.J., Darwin’s Black Box: The Biochemical Challenge to Evolution, The Free Press, New York, 1996; see review by Ury, T.H., Journal of Creation. 11(3):283–291, 1997. See also DiSilvestro, R., Rebuttals to common criticisms of the book Darwin’s Black Box (last updated 26 October 1999). Return to text.
- Denton, Ref. 4, ch. 11. Return to text.
- Jorde, L.B., Carey, J. C. and White, R.L., Medical Genetics, Mosby, St Louis, Missouri, 1995. Return to text.
Readers’ comments
Comments are automatically closed 14 days after publication.