

Journal of Creation 17(1):26–32, April 2003
Browse our latest digital issue Subscribe
Does the acquisition of antibiotic and pesticide resistance provide evidence for evolution?
The development of antibiotic and pesticide resistance is often presented as a modern example of evolution by mutations and as clear evidence for Darwinism. A literature review found that most examples of the acquisition of resistance are not due to mutations, but in nearly all cases are a result of complex, built-in genetic and molecular biological defence systems. The extant literature indicates that those few examples that are due to mutations are in all cases so far due to loss mutations and do not result in a gain of genetic information.
One of the most common arguments against the Creation worldview is the well-documented development of resistance in bacteria to antibiotics and in insects to insecticides. A typical example can be found in the book The Evolution Explosion by Harvard biologist Stephen Palumbi. In this work, according to a recent review, Palumbi discusses extensively
‘ … cases in which humans have produced rapid evolution in other species by changing their environments: his examples include the evolution of antibiotic resistance in bacteria, herbicide resistance in plants, pesticide resistance in insects, and changes in the growth rate of fish caused by overfishing. Remarkably, many people familiar with these phenomena have failed to see that they demonstrate evolution driven by selection. There is, for example, a public misconception that “drug resistance” involves not evolutionary change in pathogenic bacteria, but some process whereby a person becomes acclimated to antibiotics.’1
Many Darwinists have claimed that the development of antibiotic and pesticide-resistance is one of the strongest evidences of Darwinian evolution. Examples include Greenspan,2 Crews,3 Iltis,4 Kopaska-Merkel,5 and the PBS series Evolution.6 This paper focuses on the common claim that the development of resistance to antibiotics and insecticides provides evidence for the molecules-to-man evolution theory is based at its foundation on mutations.
Development of resistance is a major concern for another reason—human health. Infectious diseases historically have killed billions and have caused several of the most devastating chapters in the history of humankind.7 Scientists have been so successful in the past century in preventing and curing infectious diseases that only a few years ago it was thought that modern science had at last enabled us to ‘close the book on infectious diseases’.8 However, recent history has proven this conclusion to be tragically premature.9,10 Two major topics of current concern in the fields of communicable diseases are ‘emerging’ infectious diseases and ‘re-emerging’ infectious diseases such as pertussis (commonly known as whoopingcough). The development of resistance must be understood in order to deal with the serious health threat this situation causes.
Bacteria that have become resistant to several antibiotics, said to be multi-drug resistant, are often called super bugs by the media. For some pathogens vancomycin now is the only effective agent, and even it has lost some of its effectiveness in recent years.11 This problem has many causes, including the misuse of antibiotics and the transfer of resistance genes from one bacterium to another.12 This is possible because many bacteria have a built-in natural resistance to a number of antibiotics, and the genes that provide this resistance can be passed on to other bacteria by a variety of means.
Mechanisms involved in antibiotic resistance in bacteria
Non-resistant bacteria commonly become resistant by several different means, most of which have nothing to do with mutations. Palumbi notes that in ‘most cases’ antibiotic resistance results from selection of an existing genetic trait, especially those traits that are highly variable, such as the natural defences that all organisms possess.13 An important mechanism by which bacteria become resistant is by obtaining one or more specific resistance genes from other bacteria. This type of resistance can be obtained by the transfer of a plasmid (small circular units of DNA), already existing in the bacterium gene pool, that carries a gene for an enzyme which either destroys or inactivates the antimicrobial substance.14 Many resistance genes are also carried on self-transmissible genes known as transposable elements, that can jump between plasmids and chromosomes.15
Bacteria can obtain a new gene (or genes) by several methods:
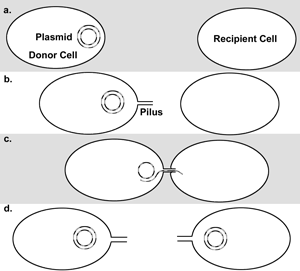
- Conjugation is the most common method. It is a complex system that transfers a copy of a plasmid from one bacterial cell (called the donor) to another bacterium (called the recipient) (Figure 1). A tube-like structure known as pilus latches onto the recipient and is positioned in such a way that a conjugation bridge can form, allowing for the transfer of genes for resistance and other purposes.16 A common example is bacterial resistance to penicillin that is acquired by obtaining the gene for penicillinase as a result of conjugation. Penicillinase is an enzyme that alters the penicillin molecule in such a way that it is rendered ineffective. The plasmid containing the drug resistance is called a Resistance (R) factor.
- Transduction is a virus-mediated transfer of host DNA from one host to another. Bacterial viruses, known as bacteriophages, sometimes can serve as intermediaries, picking up the resistance gene from a naturally resistant bacterium and then passing on this gene to non-resistant bacteria. In this case the bacteria’s genomes gain information, but the source is not mutations. Instead, the new genetic material is derived from the genome of another bacterium that already has the gene (or gene set) that confers resistance.
- Transformation (the process in which bacterium take up exogenous DNA from its environment). Chromosomal or plasmid DNA can even be taken up and spread from dead to living bacteria.
Also, many gene sets called transposons are self-transmissible and can transfer from their normal location to other plasmids or chromosomes. In bacteria, antibiotic-resistant genes are located on plasmids or transposons, small circular units of DNA that can even be spread from dead to living bacteria.
Antibiotics can be inactivated by various means
Antibiotics are produced naturally by fungi and bacteria which have coexisted since Creation as part of their own defence systems. Without innate defences, bacteria could not protect themselves and would soon become extinct. When an antibiotic reaches the bacterial periplasm or cytoplasm (see Figure 2) it may be inactivated by modification, isolation, or destruction, all of which are due not to mutations’, but rather to complex, innate, physiological mechanisms.
When a bacterial strain has gained resistance to an antibiotics, it is more correct to say that the bacteria it has lost sensitivity to the antibiotic.17 Furthermore, bacteria have had resistance to many antibiotics long before humans used them. This has been confirmed by culturing bacteria found on human explorers frozen to death long before human-developed antibiotics existed. An example is a 1988 University of Alberta study of bacteria on the bodies of Arctic explorers frozen in 1845. Investigators discovered that some of the bacterial strains were resistant to antibiotics.18 The study, which evaluated six strains of Clostridium on three men who had been buried in permafrost, found the bacteria were particularly resistance to clindamycin and cefoxitin, both antibiotics that were developed over a century after the men died.19 Clostridium is part of the normal bacteria flora of the human gut.
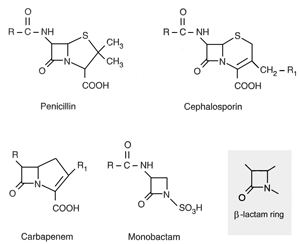
A drug also can be deactivated by modifying a critical part of its molecular structure. An example is beta-lactamase, an enzyme that attacks penicillin, primarily by destroying its β-lactam ring. As a result, the antibiotic no longer is functional, and therefore microorganisms that produce β-lactamase are resistant to all antibiotics containing the β-lactam ring (known as beta-lactam antibiotics, part of the beta-lactam family). β-lactamase is manufactured by a set of genes on R-plasmids that can be passed to other bacteria. In 1982, over 90% of all clinical staphylococcus infections were penicillinresistant, compared to close to 0% in 1952. The reason for the increase was due largely to the rapid spread (primarily by conjugation transfer) of the β-lactamase plasmid.
Some antibiotics may be effective temporarily, but cellular repair mechanisms, redundant regulatory systems, or subsequent protein synthesis later restores vitality to the bacteria. The organism may respond to the antibiotic by causing a significant increase in metabolism so that the previous level of antibiotic no longer is sufficient to interfere with the metabolic process.
Another example involves passing on a gene or genes that enable recipient bacteria to produce the compound blocked by the antibiotic. For example, sulfonamide works by blocking the bacteria’s ability to synthesize folic acid. If one or more genes coding for folic acid are acquired from other bacteria, this enables synthesis of this compound and renders the sulfonamide ineffective, or at least much less effective, providing the bacteria with sufficient folic acid for survival.
As a result of the ineffectiveness of penicillin, doctors often administer methicillin, a drug that disables another metabolic mechanism to kill bacteria. By the 1980s, several important strains of Staphylococcus also were resistant not only to methicillin, but also to another drug known as nafcillin. In 1992, almost 15% of all Staphylococcus were methicillin-resistant in the USA, and by 1993 vancomycin remained as the only antibiotic that could kill all strains of the organism.20 Staphylococci are everywhere—in the soil, on human skin, in the oral cavity—and easily can be passed on by simple body contact. The majority of the 920,000 post-surgical infections were due to staphylococcal infections, mostly by the methicillin-resistant staph strain. Strains of staphylococci that were resistant to many drugs existed naturally by 1990. A research team treated a patient infected
‘with a strain that was resistant to cadmium, penicillin, kanamycin, neomycin, streptomycin, tetracycline, and trimethoprim. Since each of these drugs operated by specific biochemical mechanisms that were used by a host of related drugs, the Australian staph could resist, to varying degrees, some thirty-one different drugs. In a series of test-tube studies the Australians showed that these various resistance capabilities were carried on different plasmids that could be separately passed from one bacterium to another. The most common mode of passage was conjugation: one bacterium simply stretched out its cytoplasm and passed plasmids to its partner.’21
A major cause for such a situation is overuse of antibiotics that select for the resistant strains, causing them to become more common.
Transporters and efflux pumps
Yet another method whereby bacteria can become resistant to antibiotics is by gaining the genes for pumps that remove the antibiotic from the cell before it can cause harm. Pumps can remove many kinds of toxins, including anti-cancer drugs. Efflux pumps use metabolic energy to remove antibiotics from the cytoplasm, thereby reducing the effective concentration of the antibiotic inside the cell. Referred to as multi-drug resistant pumps, they are produced by a number of genes (usually located on plasmids) that can be passed to other bacteria during conjugation. The pump mechanism attaches a protein label to the drug and removes it by exocytosis. A similar family of mechanisms exists in both prokaryotic and eukaryotic cells. Humans possess a superfamily of transporters such as the human P-glycoprotein, which can remove a diverse class of amphipathic (which has both hydrophobic (lipid) and hydrophylic (water attracting) regions) drugs from cells and also are a source of multi-drug resistant cancer cells.22
Resistance due to mutations
Bacteria can become resistant as a result of mutations, but all of those studied so far are loss mutations. Probably the classic example is streptomycin and other mycin drugs that have been rendered ineffective by ribosome point mutations.23,24 Mycin antibiotics function by attaching to a specific receptor site on the bacteria’s ribosomes, and thereby interfering with their protein-manufacturing process. As a result, the proteins that the bacteria produce are non-functional, so the bacteria cannot grow and divide or propagate. Mammalian ribosomes do not contain the specific site where myosin drugs can attach, and for this reason. the drug does not interfere with their ribosomes. Therefore, mycin drugs adversely affect bacterial growth without harming the host. Because fundamental differences exist between prokaryotic (bacterial) and eukaryotic ribosomes, these variations often are exploited in producing antibiotics to kill bacteria without harming the host.
Bacterial mutations cause the bacteria to become resistant to streptomycin if the ribosome site where the streptomycin attaches is altered by the mutation. As a result, the streptomycin no longer can bind, and therefore no longer interferes with the ribosomal function. Mutation-caused changes can occur in several different locations on the ribosome and still enable the bacteria to become mycin resistant.25,26 Another example of a mutation-caused resistance is found in Mycobacterium tuberculosis, which manufactures an enzyme that alters the antibiotic isoniazid into its active form, then killing the bacterium. A mutation that damages the enzyme allows the antibiotic to remain in its largely inactive and harmless form.27
Mutations that change a protein are likely to weaken the organism, and when it becomes resistant to a drug, it is likely to become less fit in other ways.28 These modifications do not improve the bacteria because they render them less able to survive in nature.29 Streptomycin-resistant bacteria actually are weaker in the wild for several reasons. The major reason is the ribosome specificity is lowered in bacteria that become resistant to streptomycin, and as a result the ribosomes’ ability to translate certain RNA transcripts into protein is less effective.30 Although reduction of binding affinity does not always result in the loss or reduction of all binding specificity, the specificity for the proteins required for efficient ribosome function is usually decreased.
Numerous empirical studies have found that mutations that confer resistance decrease the fitness of bacteria in environments without antibiotics. A result they do not reproduce as quickly as non-resistant bacteria. Evidence discovered so far indicates that these mutations render bacteria less fit in the wild because the mutant strain is less able to compete with the wild type. For example, one study compared multi-drug resistant tuberculous bacteria with non-resistant strains. It was found that the multi-drug-resistant strain had significantly decreased fitness compared to the drug-susceptible strain.31 The research also indicates that the same is true of viruses.32
Even though the mutation in this case provides the organism with a clear advantage, in the wild (i.e. an antibiotic-free environment) the change is usually not an advantage and normally would not be selected for. When the drug is no longer part of the environment, the non-resistant type is again better adapted, and the resistant type less so.28 In a relatively sterile hospital environment, however, the resistant strain has a clear advantage in those patients given antibiotics because it can render many antibiotics useless.
The classic example is a patient who develops resistance to antibiotics in a hospital, yet the infection clears when sent home because the resistant bacteria cannot compete with non-resistant normal flora. Other mutations apart from those affecting the ribosome also have been found to render bacteria streptomycin-resistant. In all of these cases, the mutation that causes the resistance results either from the degradation or loss of genetic information (from gene damage, for example, resulting in a gene product that is no longer functional).
Similar examples of natural selection also apply to humans and other creatures (i.e. resistance that results from mutations as opposed to that from natural resistance to pathogens due to normal immune system function such as from vaccination or prior exposure, such as the sickle cell anemia mutation in humans that confers resistance to malaria). However, as Schliekelman et al. note:
‘Although infectious disease is assumed to be an important cause of natural selection in humans, strong selection in favour of alleles that confer resistance to disease has been demonstrated only in the case of malaria.33
The same observation has been confirmed in other more structurally complex animals. The resistance due to mutations is evidently largely confined to viruses, bacteria and insects.
Cell surface receptors and antibiotic resistance
The outer membrane of gram-negative bacteria (a type of bacteria that has a cell wall that resists staining with certain dyes) serves as a barrier to the outside world that protects the cell. However, specific proteins (known as porins) in the outer membrane of the bacteria serve as diffusion channels or gateways for hydrophylic molecules such as certain antibiotics. Several types of porins exist, and low levels (or loss) of certain porins due to loss mutations increases the bacteria’s resistance to some antibiotics because they can no longer enter the cell, or the small amounts that enter cannot kill the cell.34 Hydrophobic molecules can diffuse through the membrane itself, but some mutations involving outer membrane biochemistry can have an adverse impact on diffusion rates; thus, these mutations potentially increase drug resistance.35–39 These mutations also evidently slow the infusion of certain nutrients and other needed materials, and as a result these resistant bacteria are normally less fit than normal bacteria in the wild.
Another mutation that may confer resistance is gene duplication, which may require a larger dose of the antibiotic to be effective if the antibiotic attacks the protein that is now produced in higher amounts.40 This mutation allows the restoration of a normal metabolic level, or at least a level that allows survival. In this case, new genes are not created, consequently this mutation is not an example of evolution. Furthermore, in normal environments the overproduction is often harmful.
Changes in the cellular target of cell surface molecules
In order to enter a bacterial cell, the drug first must bind to cell surface proteins called binding sites. Mutations cause many bacteria to become resistant to antibiotics by altering the cell surface proteins that enable the antibiotic to enter the cell. Binding and subsequent transport of antibiotics typically involve the same protein or protein complex. Some resistance may occur as a result of one or more alterations of these drug-binding sites on the cell surface so that the drug no longer can bind. It consequently is prevented or hindered from entering the cell and therefore cannot as readily accumulate to toxic levels.
Resistance also can occur as a result of alterations in membrane permeability or other changes on the cell surface that prevent the drug from binding to, and thus entering, the cell.41,42 This mutation also renders the organism less fit in nature because the damaged receptor also is less able to take in the substances it normally brings into the cell.
Another method whereby pathogens can acquire resistance is via alterations caused by mutations that in some way modify the cellular target in such a way as to render the antibiotic no longer effective. Specific trans-membrane transporters serve to import into the cell various target molecules (such as nutrients), but also may import some antibiotics such as metabolic analogues. A mutation to, or a loss of, the transporter may decrease the antibiotic level entering the cell, and consequently it will become less effective. In the case of bacteria, in order to enter a cell the drug must pass through both the cell wall and the cell membrane. If the cell membrane permeability level changes, the drug no longer is able to pass through, and as a result cannot reach its target to cause cell damage or block cell reproduction (such as by binding to DNA or to a ribosome). The organism is now resistant to this drug, but it is also less able to survive in the wild because resistance-conferring mutations in bacteria are loss mutations that render the organism less fit to survive in an antibody-free environment.
The problem of bacterial resistance does not provide evidence for evolution, but instead supports intelligent design. In no case has a mutation that results in new functional information-gain (such as one that produces a new gene) been demonstrated. The problem today is a result of several factors, including the use of antibiotics that either are not indicated, or are not given at the incorrect dosage or for the proper duration. Also, under use of prevention and vaccination are important.
One of the latest examples involves Staphylococcus aureus resistance to one of the newest modern antibiotics, vancomycin. The S. aureus builds its cell wall out of tightly crosslinked strands. A gene codes for the enzyme that constructs a ‘cap’ on the strand ends.43 This cap is used to help produce the cross linking needed to build a ‘strong tough wall that contains and protects the cell’s contents.44 Vancomycin binds to the strand end and as a result stops further cell wall formation.45 The resultant uncross linked areas are weak, allowing water to enter by osmosis. As a result the cell balloons out, causing the cell to burst and killing the bacteria. A specific mutation makes an altered enzyme that in turn produces an altered cap that is unaffected by vancomycin. In this case, the mutation clearly provides a survival advantage to the bacteria, but only in an abnormal environment. In a normal environment, though, the

‘mutant cap leads to a weaker cell wall than normal, and so populations of these mutant cells grow more slowly than normal cells. The environment now becomes key. In a regular cellular environment with no antibiotics present, staph cells with the normal gene grow quickly, cap their strand ends, crosslink them into strong cell walls, and outcompete staph cells with the mutant gene (emphasis mine).’44
Insecticide resistance
Insecticide resistance is another major problem. Some insects are tolerant to so many insecticide families that ‘chemical control is useless’.46 Developing resistance to DDT in insects functions in a similar way as streptomyosin resistance. The insecticide DDT binds to a specific matching site on the membrane of the insect’s nerve cells, interfering with the nerve cells’ functions. When a certain level of DDT binds to the nerve cells, the nervous system no longer is able to function properly, and as a result the insect dies.47 Any mutation that adversely affects the binding of DDT to the nerve cell, if it is not lethal or almost lethal, has the potential of conferring DDT resistance to the insect.48
Likewise, as is true with bacteria, insects that have become resistant to insecticides have been shown to be less fit in the wild.49 For instance, many resistant insects are less active and slower to respond to stimuli than other insects. This effect has been researched specifically in the case of mosquitoes. Although more fit in the environment in which the insecticide is present, the more sluggish nervous system results in the resistant insects being less fit in a normal insecticide-free environment. Nonetheless, prolonged use of insecticides can produce large numbers of resistant insects which, even though they are less fit as a whole, can survive better in an environment with high levels of DDT. As a result, the resistant population becomes larger in spite of its members’ overall less-effective nervous systems. The problem is so common that most insects eventually develop resistance to many insecticides:
‘Insect resistance to a pesticide was first reported in 1947 for the housefly (Musca domestica) with respect to DDT Since then the resistance to one or more pesticides has been reported in at least 225 species of insects and other arthropods. The genetic variants required for resistance to the most diverse kinds of pesticides were apparently present in every one of the populations exposed to these man-made compounds.’50
An excellent summary of the fact that pesticide resistance usually results in a clear survival disadvantage in a toxic free environment is as follows:
‘ … resistance to poisons is rarely a “free ride” for either insects or other organisms, because the selective trade-offs imposed by pleiotropy often maintain polymorphism either within or between populations of a species. Some populations of Norway rats, for example, have evolved resistance to the rat poison warfarin. Where the poison is in widespread use, homozygotes for the allele that confers resistance are common. But that allele also lowers rats’ ability to synthesize vitamin K, a compound essential in allowing blood to clot, and they bleed more easily. For that reason, in places where warfarin is not used, individuals homozygous for this allele are at as much as a 54 percent selective disadvantage compared to “wild-type” rats, and the allele is far less common. The same sort of phenomenon has been demonstrated for the alleles that confer resistance to DDT and to dieldrin in mosquitoes.’51
Another evidence that antibiotic resistance does not lend support to evolution is that the rise of antibiotic resistance as a result of mutations have been extremely rapid because the mutations need only to reduce or damage the function of pre-existing systems (i.e. reduce protein binding effectiveness, damage cell transport systems, or disrupt regulatory control). It usually requires only a single mutation (i.e. a point mutation) to reduce or eliminate a system that already is present in the cell. These mutations are easily acquired, and this is why the resulting new phenotypes are produced rapidly. Within a decade or less after a new insecticide is introduced, many insects are resistant to it. The same is true for bacteria and many other organisms. For example, DDT was discovered in 1939 but, ironically, resistance to it was reported in house flies even before its developer, Paul Muller, received the Nobel Prize for his work.52
Conversely, mutations that add new systems or information, such as a new regulatory system, a new synthetic system, a new energy-generating system, or a new transport system, have never been convincingly documented. Even Darwinists posit cons for them to occur, and they have never been shown to happen. Mutations increasing enzyme affinity are not clearly beneficial, but may occur rapidly. For example, mutations in the hemoglobin-oxygen affinity system help their hosts acclimatize to high altitude but they also cause polycythemia (an increase in the red blood cell count or the concentration of hemoglobin in the red blood cells as a means of adapting to the low level of oxygen).
Conclusion
The recent development of bacterial and insect resistance does not support neo-Darwinism classically defined as the natural selection of mutations. Evolution requires information-building mechanisms that add new information to DNA. In virtually all cases, bacteria or insect resistance is a result of the exploitation of existing systems, or is due to a transfer of genes. In the rare cases where a mutation is involved, development of resistance involves only a loss mutation such as one that produces a deformed ribosome. This is confirmed by the fact that resistance is acquired very rapidly, in far too brief a period for the evolutionary emergence of complex biochemical or physiological systems. Mutation caused resistance results in less viability in the wild, and as a result the resistant stains cannot compete.
The multi-drug resistance problem is not small—-and now results in tens of thousands of deaths annually. Human uses and abuses are the major cause, not Darwinian evolution. The acquisition of antibiotic resistance does not provide evidence for microbe-to-man evolution but rather for intelligent design and only by understanding the mechanism involved can the resistance problem be solved.53
Acknowledgments
I wish to thank Kevin Anderson, David Demick, Bert Thompson, John Woodmorappe, Clifford Lillo and Wayne Frair for their comments on an earlier draft of this manuscript.
References
- Coyne, J., The case of the missing carpaccio, Nature 412:586–587, 2001; p. 586. Return to text.
- Greenspan, N.S., Not-so-intelligent design, The Scientist 16(5):12, 2002. Return to text.
- Crews, F.C., Saving us from Darwin, The New York Review of Books, 4 October 2001. Return to text.
- Iltis, E., Letters to the Editor, Reports of the National Center for Science Education 20(5):44–45, 2000. Return to text.
- Kopaska-Mentel, D., Letters to the Editor, Reports of the Natural Center for Science Education 20(5):44, 2000. Return to text.
- Gilder, J., Review of evolution,http://worldnetdaily.com, 2001. Return to text.
- Marks, G. and Beatty, W.K., Epidemics, Charles Scribner’s Sons, New York, 1976. Return to text.
- Berkelman, R. and Hughes, J., Editorial: The conquest of infectious diseases: who are we kidding? Annals of Internal Medicine 119(5):426–427, 1993; p. 426. Return to text.
- Rothman, K.J. and Greenland, S., Modern Epidemiology, 2nd Edition, Lippincott-Raven Publishers, Philadelphia, 1998. Return to text.
- Garrett, L., The Coming Plague: Newly Emerging Diseases in a World Out of Balance, Farrar, Straus, and Giroux, New York, 1994. Return to text.
- Padiglione, A.A., Grabsch, E.A., Olden, D., Hellard, M., Sinclair, M.I., Fairly, C.K. and Grayson, M.L., Fecal colonization with vancomycin-resistant enterococci in Austria, Emerging Infectious Diseases 6(5):534–536, 2000. Return to text.
- Finegold, S.M., Antimicrobial therapy of anaerobic infections: a status report, Hospital Practice 14(10):71–81, 1979. Return to text.
- Palumbi, S.R., Evolution—humans as the world’s greatest evolutionary force, Science 293:1786–1790, 2001; p. 1787. Return to text.
- Burton, G. and Engelkirk, R, Microbiology for the Health Sciences, Lippincott Williams and Wilkins, Philadelphia, pp. 199–201, 2000. Return to text.
- Walkup, L., Junk DNA, Journal of Creation. 14(2):18–30, 2000. Return to text.
- Black, L, Microbiology: Principles and Explorations, John Wiley and Sons, New York, 2002. Return to text.
- Rowland, M., Fitness of insecticide resistance, Nature 237:194, 1987. Return to text.
- McGuire, R., Eerie: human arctic fossils yield resistant bacteria, Medical Tribune, 29 December, 1988, pp. 1, 23. Return to text.
- Struzik, E., Ancient bacteria revived, Sunday Herald, 16 September 1990, P. 1. Return to text.
- Garrett, Ref. 10, p. 412. Return to text.
- Garrett, Ref. 10, p. 413. Return to text.
- Chang, G. and Roth, C.B., Research article—Structure of MsbA from E. coli: a homolog of the multidrug resistance ATP binding cassette (ABC) transporters, Science 293:1793–1800, 2001. Return to text.
- Davies, L, Brzezinska, M. and Benveniste, R., R factors: biochemical mechanisms of resistance to amino glycoside antibiotics, Annals of the New York Academy of Science 182:226–233, 1971. Return to text.
- Davies, J. and Nomura, M., The genetics of bacterial ribosomes, Annual Review of Genetics 6:203–234, 1972. Return to text.
- Didier, E. S., Bertucci, D.C. and Leblanc, L., Inhibition of microsporidia growth in vitro, Abstracts of the General Meeting American Society Microbiology 99:11, 1999. Return to text.
- Kazanjian, P., Armstrong, V., Hossler, P.A., Burman, V., Richardson, L, Lee, C.H., Crane, L., Katz, J. and Meshnick, S.R., Pneumocystis carinii mutations are associated with duration of sulfa or sulfone prophylaxis exposure in AIDS patients, J. Infectious Diseases 182(2):551–557, 2000. Return to text.
- Wieland, C., Antibiotic resistance in bacteria, Journal of Creation 8(l):5–6, 1994; p. 5. Return to text.
- Spetner, L., Not By Chance, The Judaica Press, Brooklyn, p. 144, 1997. Return to text.
- Wieland, Ref. 27, p. 6. Return to text.
- Gartner, T. and Orias, E., Effects of mutations to streptomycin resistance on the rate of translation of mutant genetic information, J. Bacteriology 91:1021–1028, 1966. Return to text.
- Davies, A.P., Billington, O.J., Bannister, B.A., Weir, W.R., McHugh, T.D. and Gillespie, S.H., Comparison of fitness of two isolates of mycobacterium tuberculosis, one of which had developed multi-drug resistance during the course of treatment, J. Injection 41(2):184–187, 2000. Return to text.
- Martinez-Picado, L, Savara, AY, Sutton, L. and ‘D’Aquila, R.T., Replicative fitness of protease inhibitor-resistant mutants of human immunodeficiency virus type 1, J. Virology 73(5):3744–3752, 1999. Return to text.
- Schliekelman, P., Garner, C. and Slatkin, M., Natural selection and resistance to HIV; a genotype that lowers susceptibility to HIV extends survival at a time of peak fertility, Nature 411:545, 2001. Return to text.
- Penrose, E., Bacterial resistance to antibiotics—a case of un-natural selection, CRSQ 35:76–82, 1998. Return to text.
- Chevalier, L, Pages, J.M., Eyraud, A. and Mallea, M., Membrane permeability modifications are involved in antibiotic resistance in Klebsiella pneumoniae, Biochemical and Biophysical Research Communications 274(2):496–499, 2000. Return to text.
- De, E., Basle, A., Jaquinod, M., Saint, N., Mallea, M., Molle, G. and Pages, J.M., A new mechanism of antibiotic resistance in Enterobacteriaceae induced by a structural modification of the major porin, Molecular Microbiology 41(l):189–198, 2001. Return to text.
- Domenech-Sanchez, A., Hemandez-Alles, S., Martinez-Martinez, L., Benedi, V.J. and Alberti, S., Identification and characterization of a new porin gene of Klebsiella pneumoniae: Its role in beta-lactam antibiotic resistance, J. Bacteriology 181(9):2726–2732, 1999. Return to text.
- Wong, K.K., Brinkman, F.S., Benz, R.S. and Hancock, R.E., Evaluation of a structural model of Pseudomonas aeruginosa outer membrane protein OprM, an efflux component involved in intrinsic antibiotic resistance, J. Bacteriology 183(1):367–374, 2001. Return to text.
- Cornaglia, G., Mazzariol, A. and Fontana, R., The astonishing complexity of antibiotic resistance, Clin. Microbiol. Infect. 6(Suppl 3):93–94, 2000. Return to text.
- Penrose, Ref. 34, p. 78. Return to text.
- Cohen, M.L., Epidemiology of drug resistance: implications for a post-antimicrobial era, Science 257:1050–1055, 1992. Return to text.
- Gentry, L.O., Bacterial resistance, Orthopedic Clinics of North America 22:379–388, 1991. Return to text.
- Yan, H., Zhao, Q, Yuan, J., Cheng, X. and He, B., Affinity adsorbents for the Vancomycin Group of antibiotics, Biotechnology and Applied Biochemistry 31(Pt 1):15–20, 2000. Return to text.
- Postlethwait, J.H. and Hopson, J.L., Explore Life, Brooks/Cole Thomson Learning, Australia, p. 220, 2003. Return to text.
- Mulichak, A.M., Losey, H.C., Walsh, C.T. and Garavito, R.M., Structure of the UDP-Glucosyltransferase GtfB that modifies the Heptapeptide Aglycone in the Biosynthesis of Vancomycin Group Antibiotics, Structure 9(7):547–557, 2001. Return to text.
- Palumbi, Ref. 13, p. 1786. Return to text.
- Beeman, R., Recent advances in mode of action of insecticides, Annual Review of Entomology 27:253–281, 1982. Return to text.
- Tananka K., Scott J. and Matsumura, E, Picrotoxinin receptor in the central nervous system of the American cockroach; its role in the action of cyclodiene-type insecticides, Pesticide Biochemistry and Physiology 22:117–127, 1984. Return to text.
- Panlilio et al. and Toner, M., When bugs fight back; Pulitzer Prize-winning series of reports in the Atlanta Constitution, 23 August–16 October 1992. Return to text.
- Ayala, E, The mechanisms of evolution, Scientific American 239(3):56–69, 1978; p. 65. Return to text.
- Levine, J. and Miller, K., Biology: Discovering Life, D.C. Heath, Lexington, p. 257, 1994. Return to text.
- Palumbi, Ref. 13, p. 1786. Return to text.
- Thompson, B., Bacterial antibiotic resistance—proof of evolution? Reason and Revelation 14(8):61–63, 1994. Return to text.
Readers’ comments
Comments are automatically closed 14 days after publication.