

Journal of Creation 22(2):79–84, August 2008
Browse our latest digital issue Subscribe
Evidence for the design of life: part 1—Genetic redundancy
Knockout strategies have demonstrated that the function of many genes cannot be studied by disrupting them in model organisms because the inactivation of these genes does not lead to a phenotypic effect. For living systems, this peculiar phenomenon of genetic redundancy seems to be the rule rather than the exception. Genetic redundancy is now defined as the situation in which the disruption of a gene is selectively neutral. Biology shows us that 1) two or more genes in an organism can often substitute for each other, 2) some genes are just there in a silent state. Inactivation of such redundant genes does not jeopardize the individual’s reproductive success and has no effect on survival of the species. Genetic redundancy is the big surprise of modern biology. Because there is no association between redundant genes and genetic duplications, and because redundant genes do not mutate faster than essential genes, redundancy therefore brings down more than one pillar of contemporary evolutionary thinking.
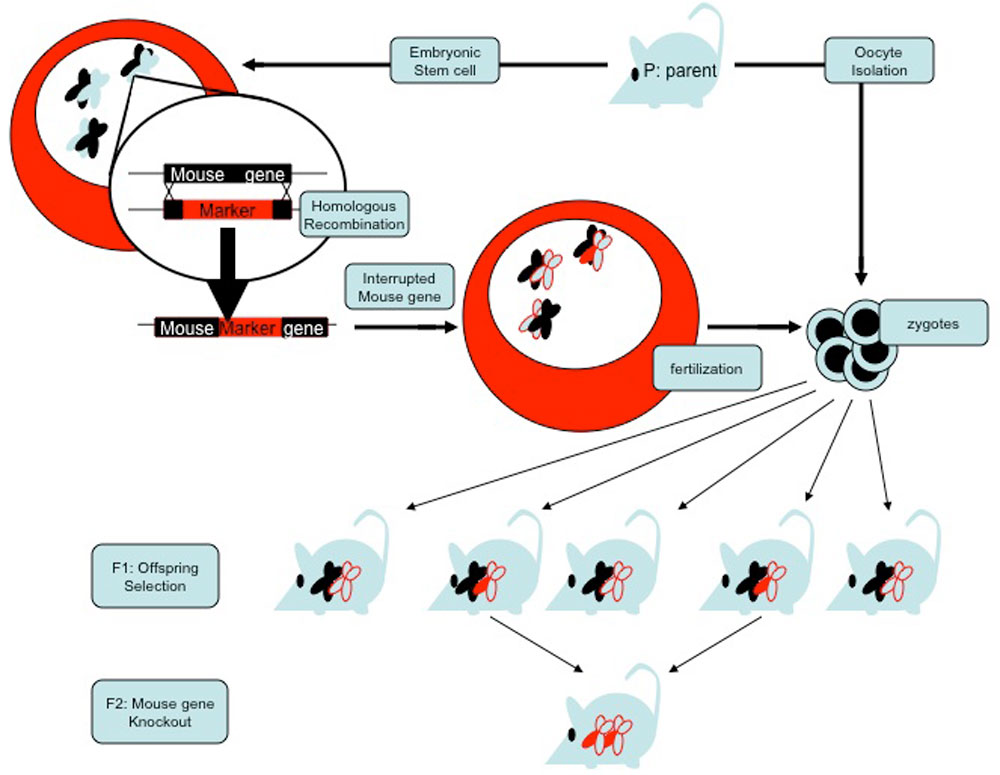
The discovery of the primary rules governing biology in the second half of the 20th century paved the way for a more fundamental understanding of the complexity of life. One of the spin-offs of this knowledge has been the development of sophisticated techniques to elucidate the function of proteins. When molecular biologists want to know the function of a particular human protein they genetically modify a laboratory mouse so that it lacks the corresponding gene (for the laboratory procedure see figure 1). Mice that have both alleles of a gene interrupted cannot produce the corresponding protein—they are called knockouts. Theoretically, the phenotype of a mouse lacking specific genetic information could provide essential information about the function of the gene. Over the years, thousands of knockouts have been generated. The knockout-strategy has helped elucidate the functions of hundreds of genes and has contributed immensely to our biological knowledge. However, there has been one unexpected surprise—the no-phenotype knockout. This is unexpected, because according to the Darwinian paradigm, all genes should have a selectable advantage. Hence, knockouts should have measurable, detectable phenotypes. The no-phenotype knockouts demonstrate that genes can be disrupted without—or with only minor—detectable effects on the phenotype. Many genes seem to have no measurable function! This is known as genetic redundancy and it is one of the big surprises of modern biology.
Molecular switches
One of the most intriguing examples of genetic redundancy is found in the SRC gene family. This family comprises a group of eight genes that code for eight distinct proteins all with a function that is technically known as tyrosine kinase. SRC proteins attach phosphate groups to other proteins that contain the amino acid tyrosine in a specific amino acid context. The result of this attachment is that the protein becomes activated; it is switched on, and can hence pass down information in a signalling cascade. Four closely related members of the family are named SRC, YES, FYN and FGR, and the other related members are known as BLK, HCK, LCK and LYN. Both families are so-called ‘nuclear receptors’, and transmit signals from the exterior of the cell to the nucleus, the operation centre where the information present in the genes is transcribed into messenger RNA. The proteins of the SRC gene family operate as molecular switches that regulate growth and differentiation of cells. When a cell is triggered to proliferate, tyrosine kinase proteins are transiently switched on, and then immediately switched off.
The SRC gene family is among the most notorious genes known to man, since they cause cancer as a consequence of single point mutations. A point mutation is a change in a DNA sequence that alters only one single nucleotide—a DNA letter—of the entire gene. When the point mutation is not on a silent position, it will cause the organism’s protein-making machines to incorporate a wrong amino acid. The consequence of the point mutation is that the organism now produces a protein that cannot be switched off. Mutated SRC genes are of particular danger because they will permanently activate signalling cascades that induce cell proliferation: the signal that tells cells to divide is permanently switched on. The result is uncontrolled proliferation of cells—cancer. The growth-promoting point mutations cannot be overcome by allelic compensation because a normal protein cannot help to switch off the mutated protein.
Despite the SRC protein being expressed in many tissues and cell types, mice in which the SRC gene has been knocked out are still viable. The only obvious characteristic of the knockout is the absence of two front teeth due to osteoporosis. In contrast, there are essentially no point mutations allowed in the SRC protein without severe phenotypic consequences. Amino acid changing point mutations in most, presumably all, of the SRC genes can lead to uncontrolled cellular replication.1 Knockout mice models have been generated to reveal the functions of all the members of the SRC gene family. Four out of eight knockouts did not have a detectable phenotype. Despite their cancer-inducing properties, half of the SRC genes appear to be redundant. Standard evolutionary theory tells us that redundant gene family members originated through gene duplications. Duplicated genes are truly redundant and as such they are expected to reduce to a single functional copy over time through the accumulation of mutations that damage the duplicated genes. Such mutations can be frame-shift mutations that introduce premature stop signals, which are recognized by the cellular translation-machines to terminate protein synthesis. The existence of the SRC gene family has been explained as follows:
‘In the redundant gene family of SRC-like proteins, many, perhaps almost all point mutations that damage the protein also cause deleterious phenotypes and kill the organism. The genetic redundancy cannot decay away through the accumulation of point mutations.’1
This scenario implies that the SRC genes are destined to reside in the genome forever. Point mutations that immediately kill raise an intriguing origin question. If the SRC genes are really so potently harmful that point mutations induce cancer, how could this extended gene family come into existence through gene duplication and diversify through mutations in the first place? After the first duplication, neither of the genes is allowed to change because it will invoke a lethal phenotype and kill the organism through cancer. Amino acid changing mutations in the SRC genes will permanently be selected against. The same holds true for the third, fourth and additional gene duplication. New gene copies are only allowed to mutate at neutral sites that do not replace amino acid in the protein. Otherwise the organism will die from tumours. Because of this ‘purifying’ selection mechanism, the duplicates should remain as they are. Yet the proteins of the SRC family are distinctly different, only sharing 60–80% of their sequences.
Redundancy—the rule not the exception
In 1964, a ‘knockout’ cross-country skier won two gold medals during the Winter Olympics in Innsbruck. In true Olympic tradition, Eero Maentyranta’s 15 and 30 km success was surrounded by controversy. Tests showed that he had 15% more red blood cells than normal subjects and Eero was accused of using doping to increase his level of red blood cells. Yet no trace of blood doping could be found. In 1964 nobody knew, but modern biology showed Maentyranta had a mutated EPO gene, which codes for erythropoietin, a messenger protein that tells the bone marrow to increase the production of red blood cells. To increase red blood levels, EPO binds to the EPO receptor that generates two opposite signals: one to instruct bone marrow cells to become red blood cells (the on-switch) and one to reduce production of red blood cells (the off-switch). This auto-regulatory mechanism assures a balanced production of red blood cells. In 1993, it turned out that the Olympic medallist had a mutation that knocked out the off-switch.2 The EPO receptor of the Finnish athlete generated a normal activation signal, but not the deactivating one. People can do well without the off-switch.
In humans, the muscle-fiber-producing ACTN3 gene can also be missed entirely and without consequences for fitness.3 Humans can also do without the GULO gene,4 the gene coding for caspase 12,5 the CCR5 gene6 and some of the GST genes that are involved in the detoxification of polycyclic aromatic hydrocarbons present in cigarette smoke.7 All these genes can be found inactivated in entire human populations (GULO, caspase 12) or subpopulations thereof. The Douc Langur (Pygathrix nemaeus), an Asian leaf-eating Colobine monkey, is the natural no-phenotype knockout for the angiogenin gene that codes a small protein that stimulates the formation of blood vessels.8 Bacterial genomes can be reduced by over 9% without selective disadvantages on minimal medium,9 and mice in which 3 megabases of conserved DNA was erased showed no signs of reduced survival and there was no indication of overt pathology.10 Fewer than 2% of approximately 200 Arabidopsis thaliana (Mouse-Ear Cress) knockouts displayed significant phenotypic alterations. Many of the knockouts did not affect plant morphology even in the presence of severe physiological defects.11 In the nematode worm Caenorhabditis elegans a surprising 89% of single-copy and 96% of duplicate genes show no detectable phenotypic effect when they are knocked out.12 Prion proteins are thought to have a function in learning processes, but when they are misfolded they can cause bovine spongiform encephalitis (BSE) or Kreutzfeld–Jacob disease. In order to make BSE resistant cows, a knockout breed has been created lacking the prion protein. A thorough health assessment of this knockout breed revealed only small differences from wild-type animals. Apparently, cows can thrive very well without the prion protein.13 Research on histone H1 genes, once believed to be indispensable for DNA condensation, suggest that any individual H1 subtype is not necessary for mouse development, and that loss of even two subtypes is tolerated if a normal H1-to-nucleosome stoichiometry is maintained.14 Even complete highly specialized cells can be redundant. A strain of laboratory mouse, named WBB6F1, lacks a specific type of blood cells known as mast cells. The reported no-phenotype knockouts are probably only the tip of the iceberg. As reported in Nature below, few knockout organisms in which no phenotype could be traced ever see the light of day:
‘ … a lot of those things [no-phenotype knockouts] you don’t hear about. No-phenotype knockouts are negative results, and as such they are usually not reported in scientific journals; because they do not have news value. To address the problem, the journal Molecular and Cellular Biology has since 1999 a section given over to knockout and other mutant mice that seem perfectly normal.’15
So how are genes, cells and organisms supposed to have evolved without selective constraints? If organisms can do without complete cells, it would be outlandish to assert that natural selection was the driving force shaping those cells. Two decades of knockout experiments has made it clear that genetic redundancy is a major characteristic of all studied life forms.
Paradigm lost
Genetic redundancy falsifies several evolutionary hypotheses. Firstly, truly redundant genes are impossible paradoxes because natural selection cannot prevent the accumulation of harmful mutations in these genes. Hence, natural selection cannot prevent redundancies from being lost. Secondly, redundant genes do not evolve (mutate) any faster than essential genes. If protein evolution is due in large part to neutral and slightly deleterious amino acid substitutions, then the incidence of such mutations should be greater in proteins that contribute less to individual reproductive success. The rationale for this prediction is that non-essential proteins should be subject to weaker purifying selection and should accumulate mildly deleterious substitutions more rapidly. This argument, which was presented over twenty years ago, is fundamental to many theoretical applications of evolutionary theory, but despite intense scientific scrutiny the prediction has not been confirmed. In contrast, a systematic analysis of mouse genes has shown that essential genes do not evolve more slowly than non-essential ones.16 Likewise, E. coli proteins that operate in huge redundant networks can tolerate just as many mutations as unique single-copy proteins,17 and scientists comparing the human and chimpanzee genomes found that non-functional pseudogenes, which can be considered as redundancies, have similar percentages of nucleotide substitutions as do essential protein-coding genes.18 Thirdly, as discussed in more detail below, several recent biology studies have provided evidence that genetic redundancy is not associated with gene duplications.
What does the evolutionary paradigm say?
An important question that needs to be addressed is—can we understand genetic redundancy from Darwin’s natural selection perspective? How can genetic redundancy be maintained in the genome without natural selection acting upon it continually? How did organisms evolve genes that are not subject to natural selection? First, let’s look at how it is thought genetic redundancies arise. Susumo Ohno’s influential 1970 book, Evolution by Gene Duplication, deals with this idea.19 Sometimes, during cell divisions, a gene or longer stretch of biological information is duplicated. If duplication occurs in germ line cells and become inheritable, the exact same gene may be present twofold in the genome of the offspring—a genetic back-up. Ohno argues that gene and genome duplications are the principal forces that drive the increasing complexity of Darwinian evolution, referring to the evolution from microbes to microbiologists. He proposes that duplications of genetic material provide genetic redundancies which are then free to accumulate mutations and adopt novel biological functions. Duplicated DNA elements are not subject to natural selection and are free to transform into novel genes. With time, he argues, a duplicated gene will diverge with respect to expression characteristics or function due to accumulated (point) mutations in the regulatory and coding segments of the duplicate. Duplicates transforming into novel genes with a selective advantage will certainly be favored by natural selection. Meanwhile, the genetic redundancy will protect old functions as new ones arise, hence reducing the lethality of mutations. Ohno estimates that for every novel gene to arise through duplication, about ten redundant copies must join the ranks of functionless DNA base sequence.20 Diversification of duplicated genetic material is now the accepted standard evolutionary idea on how genomes gain useful information. Ohno’s idea of evolution through duplication also provides an explanation for the no-phenotype knockouts: if genes duplicate fairly often, it is then reasonable to expect some level of redundancy in most genomes, because duplicates provide an organism with back-up genes. As long as duplicates do not change too much, they may substitute for each other. If one is lost, or inactivated, the other one takes over. Hence, Ohno’s theory predicts an association between genetic redundancy and gene duplication.
The evolutionary paradigm is wrong

Some biologists have looked into this matter specifically using the wealth of genetic data available for Saccharomyces cerevisiae—the common baker’s yeast. A surprising 60% of Saccharomyces’ genes could be inactivated without producing a phenotype. In 1999, Winzeler and co-workers reported in Science that only 9% of the non-essential genes of Saccharomyces have sequence similarities with other genes present in the yeast’s genome and could thus be the result of duplication events.21 Most redundant genes of Saccharomyces are not related to genes in the yeast’s genome, which suggests that genetic duplications cannot explain genetic redundancy. In 2000, Andreas Wagner confirmed Winzeler’s original findings that weak or no-effect (i.e. non-essential and redundant) genes are no more likely to have paralogous—that is, duplicated—genes within the yeast genome than genes that do result in a defined phenotype when they are knocked out. Wagner concluded that the robustness of mutant strains cannot be caused by gene duplication and redundancy, but is more likely due to the interactions between unrelated genes.22 More recent studies have shown that cooperating networks of unrelated genes contribute significantly more to robustness than gene copy number.23 Redundant genes are proposed to have originated in gene duplications, but we do not find a link between genetic redundancy, and duplicated genes in the genomes. Gene duplication is not a major contributor to genetic redundancy, and the robust genetic networks found in organisms cannot be explained. The predicted association between genetic redundancy and gene duplication is non-existent. Ohno’s interesting idea of evolution by gene duplication therefore cannot be right.
The non-linearity of biology
The no-phenotype knockouts can only be explained by taking into account the non-linearity of biochemical systems. It is ironic that standard wall charts of biochemical reactions show hundreds of coupled reactions working together in networks, while graduate students are tacitly encouraged to think in terms of linear cause and effect. The linear cause-and-effect thinking in ancient Greek philosophy was adopted by nineteenth century European scholars, and is still dominating most fields of science, including biology. We cannot understand that genetic redundancy and biological robustness in linear terms of single causality, where A causes B causes C causes D causes E. Biological systems do not work like that. Biological systems are designed as redundant scale-free networks. In a scale-free network the distribution of node linkage follows a power law in that it contains many nodes with a low number of links, few nodes with many links and very few nodes with a high number of links. A scale-free network is very much like the Golden Orb’s web: individual nodes are not essential for letting the system function as a whole. The internet is another example of a robust scale-free network: the major part of the websites makes only a few links, a lesser fraction make an intermediate number of links, and a minor part makes the majority of links. Usually hundreds of routers routinely malfunction on the Internet at any moment, but the network rarely suffers major disruptions. As many as 80% of randomly selected Internet routers can fail, but the remaining ones will still form a compact cluster in which there will still be a path between any two nodes.24 Likewise, we rarely notice the consequences of thousands of errors that routinely occur in our cells.
Scale free networks
Genes never operate alone but in redundant scale-free networks with an incredible level of buffering capacity. In a simple non-linear biological system—presented in figure 2—with nodes A through E, A may cause B, but A also causes D independent of B and C. This very simple network of only five nodes demonstrates robustness due to redundancy of B and C. If A fails to make the link with D, there are still B and C to make the connection. Extended networks composed of hundreds of interconnected proteins ensure that if one network becomes inactivated by a mutation, essential pathways will then not be shut down immediately. A network of cooperating proteins that can substitute for or bypass each other’s functions makes a biological system robust. It is hard to imagine how selection acts on individual nodes of a scale-free, redundant system. Complex engineered systems rely on scale-free networks that can incorporate small failures in order to prevent larger failures. In a sense, cooperating scale-free networks provide systems with an anti-chaos module which is required for stability and strength. Scale-free genetic and protein networks are an intrinsic, engineered characteristic of genomes and may explain why genetic redundancy is so widespread among organisms. Genetic networks usually serve to stabilize and fine-tune the complex regulatory mechanisms of living systems. They control homeostasis, regulate the maintenance of genomes and provide regulatory feedback on gene expression. An overlap in the functions of proteins also ensures that a cell does not have to respond with only ‘on’ or ‘off’ in a particular biochemical process, but instead may operate somewhere in between.
Most genes in the human genome are involved in regulatory networks that detect and process information in order to keep the cell informed about its environment. The proteins operating in these networks come as large gene families with overlapping functions. In a cascade of activation and deactivation of signalling proteins, external messages are transported to the nucleus with information about what is going on outside so it can respond adequately. If one of the interactions disappears, this will not immediately disturb the balance of life. The buffering capacity present in redundant genetic networks also provides the robustness that allows living systems to propagate in time. In a linear system, one detrimental mutation would immediately disable the system as a whole: the strength of a chain is determined by its weakest link. Interacting biological networks, where parallel and converging links independently convey the same or similar information, almost never fail. The Golden Orb’s web only crumbles when an entire spoke is obliterated in a crash with a Dragonfly, an event that will hardly ever happen. Biological systems operate as a spider’s web: many interacting and interwoven nodes produce robust genetic networks and are responsible for genetic redundancy.23
Conclusion
Genetic redundancy is an amazing property of genomes and has only recently become evident as a result of negative knockout experiments. Protein-coding genes and highly conserved regions can be eliminated from the genome of model organisms without a detectable effect on fitness. There is no association between redundant genes and gene duplications, and redundant genes do not mutate faster than essential genes. Genetic redundancy stands as an unequivocal challenge to the standard evolutionary paradigm, as it questions the importance of Darwin’s selection mechanism as a major force in the evolution of genes. It is also important to realize that redundant genes cannot have resided in the genome for millions of years, because natural selection, a conservative force, cannot prevent their destruction due to debilitating mutations. Mainstream biologists who are educated in the Darwinian framework are unable to understand the existence of genes without natural selection. This is clear from a statement in Nature a few years ago by Mario Cappecchi, a pioneer in the development of knockout technology:
‘I don’t believe that there is a single [knockout] mouse that does not have a phenotype. We just aren’t asking the right questions.’15
The right question to be asked is: is the evolutionary paradigm wrong? My answer is yes, it is. Current naturalistic theories do not explain what scientists observe in the genomes. Genetic redundancy is the actual key to help us understand the robustness of organisms and also their built-in flexibility to rapidly adapt to different environments. In part 2 of this series of articles, I will explain genetic redundancy in the context of baranomes, the multipurpose genomes baramins were originally designed with in order to rapidly spread to all the corners and crevices of the earth.
References
- Toby, J.G. and Spring, J., Genetic redundancy in vertebrates: polyploidy and persistence of genes encoding multidomain proteins, Trends in Genet. 14:46–49, 1998. Return to text.
- de la Chapelle, A., Sistonen, P., Lehvaslaiho, H., Ikkala, E. and Juvonen, E., Familial erythrocytosis genetically linked to erythropoietin receptor gene, Lancet 341:82–84,1993. Return to text.
- North, K.N., Yang, N., Wattanasirichaigoon, D., Mills, M., Easteal, S. and Beggs, A.H., A common non-sense mutation results in alpha-actinin 3 deficiency in the general population: evidence for genetic redundancy in humans, Nature Genet. 21:353–354, 1999. Return to text.
- Truman, R. and Terborg, P., Why the shared mutations in the hominidae exon X GULO pseudogene are not evidence for common descent, J. Creation 21(3):118–127, 2007. Return to text.
- The Chimpanzee Sequencing and Analysis Consortium, Initial sequence of the chimpanzee genome and comparison with the human genome, Nature 437:69–87, 2005. Return to text.
- Galvani, A.P. and Novembre, J., The evolutionary history of the CCR5-Delta32 HIV-resistance mutation, Microbes Infect. 7:302–309, 2005. Return to text.
- in: Van Diemen, C., Genetics of Lung Function Decline and COPD
evelopment, Ph.D. dissertation, Gildeprint Enschede, Netherlands, p. 127, 2008. Return to text. - Zhang, J. and Zhang, Y.P., Pseudogenization of the tumor-growth promoter angiogenin in a leaf-eating monkey, Gene 308:95–101, 2003. Return to text.
- Kolisnychenko, V., Plunkett, G., Herring, C.D., Feher, T., Posfai, J., Blattner, F.R. and Posfai, G., Engeneering a reduced Escherichia coli genome, Genome Res. 12:640–647, 2002. Return to text.
- Pennisi, E., The biology of genomes meeting: disposable DNA puzzles researchers, Science 304:1590–1591, 2004. Return to text.
- Bouche, N. and Bouchez, D., Arabidopsis gene knockout: phenotypes wanted, Curr. Opin. Plant. Biol. 4:111–117, 2001. Return to text.
- Conant, G.C. and Wagner, A., Duplicate genes and robustness to transient gene knock-downs in Caenorhabditis elegans, Proc. Biol. Sci. 27: 89–96, 2004. Return to text.
- Richt, J.A. et al., Production of cattle lacking the prion protein, Nature Biotech. 25:132–138, 2007. Return to text.
- Fan, Y., Sirotkin, A., Russell, R.G., Ayalla, J. and Scoultchi, A.I., Individual somatic H1 subtypes are dispensable for mouse development even in mice lacking the H1(0) replacement subtype, Mol. Cell. Biol. 21:7933–7943, 2001. Return to text.
- Quoted from: Pearson, H., Surviving a knockout blow, Nature 415:8–9, 2002. Return to text.
- Hurst, L.D. and Smith, N.G.C., Do essential genes evolve slowly? Curr. Biol. 9:747–750, 1999. Return to text.
- Hahn, M.W, Conant, G.C. and Wagner, A., Molecular evolution in large genetic networks: does connectivity equal constraint? J. Mol. Biol. 58:203–211, 2004. Return to text.
- Nachman, M.W. and Crowell, S.L., Estimate of the mutation rate per nucleotide in humans, Genetics 156:297–304, 2000. Return to text.
- Ohno, S., Evolution by Gene Duplication, Springer, New York, 1970. Return to text.
- Ohno, S., Evolutional reason for having so much junk DNA; in: Modern Aspects of Cytogenetics: Constitutive Heterochromatin in Man, Pfeiffer, R.A. (Ed.), F.K. Schattauer Verlag, Stuttgart, Germany, pp. 169–173, 1973. Return to text.
- Winzeler, E.A. et al., Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis, Science 285: 901–906, 1999. Return to text.
- Wagner, A., Robustness against mutations in genetic networks of yeast, Nat. Genet. 24:355–361, 2000. Return to text.
- Kitami, T. and Nadeau, J.H., Biochemical networking contributes more to genetic buffering in human and mouse metabolic pathways than does gene duplication, Nat. Genet. 32:191–194, 2002. Return to text.
- Barabasi, A. and Bonabeau, L.E., Scale-free networks, Sci. Am. 288:60–69, 2003. Return to text.


Readers’ comments
Comments are automatically closed 14 days after publication.