

Journal of Creation 23(1):70–77, April 2009
Browse our latest digital issue Subscribe
The Neutral Model of evolution and recent African origins
The Recent African Origins (RAO) theory is on tenuous ground. It relies deeply on the Standard Neutral Model of evolution (SNM), but every assumption behind SNM and, therefore, RAO has been openly questioned in the evolutionary literature. If real-world populations violate the central assumptions of SNM, the conclusions of studies that assume SNM are not the final word on the subject. The real situation is much more complicated than the simplifying assumptions allow, and several of these assumptions are either biased in favour of the conclusion or are contrary to the data: crossing over is not random, population structure exists at all scales, and population admixture and gene conversion overly complicate the models. The presence of natural selection among human mitochondria removes the ‘neutral’ part of SNM as far as RAO’s ‘Mitochondrial Eve’ is concerned. Finally, RAO and SNM are based on the belief that evolution can occur, but that it cannot affect the things that control the speed of evolution. There is no room for differences in mutation rates among populations caused by environmental stress, nutrition, demographics or mutations in the DNA copying, proofreading or correcting mechanisms. The most popular evolutionary model of recent human evolution is unsatisfactory, but the biblical model for human genetic history is still in its infancy. Outlined are several lines of thought that may be productive to creationist research.
‘The challenge of genetic studies of human history is to use the small amount of genetic differentiation among populations to infer the history of human migrations’ (Rosenberg et al. 20021).

This quote illustrates an important point. Modern geneticists are struggling to understand human genetic history. In the end, they are forced to make certain inferences based on limited data and a suite of simplifying assumptions. The purpose of this article is to look at the underpinnings of the Recent African Origins (RAO) model of human evolution, first popularized by claims of the discovery of ‘Mitochondrial Eve’ in Africa.2 Each of the fundamental assumptions behind the theory has been openly questioned in the evolutionary literature. If any one of the assumptions behind RAO falls, the entire theory may be made moot. By listing the assumptions and then systematically showing how each one is impractical, impossible, contradictory or biased in favour of evolutionary theory, I hope to bring RAO down a few notches.
The term ‘recent’ is used by RAO supporters in a deep-time sense and is not meant, by them, to be taken as support of a young Earth. According to RAO, humans evolved in Africa, existed as a small population for some hundreds of thousands of years, and then rapidly expanded into the rest of the world about 200,000 years ago. As an explanatory tool, it stands in direct opposition to the biblical model, where the most important genetic signals should be the Creation (which limits overall human diversity), the Flood (a bottleneck event), and the Tower of Babel event (which led to significant population subdivision and a world-wide migration). The last two are expected to yield similar results to the hypothetical RAO model, but with a different timescale.
I will caution the reader at this point. This article might seem to overthrow all arguments based on neutral evolution or bottleneck theories (both terms will be defined below). Creationists sometimes use those theories to their advantage, and it is not my intent to completely discount them. In fact, RAO uses much of the same mathematics many creationists would like to apply to the biblical model. My intent is to take a more ‘surgical’ approach, cutting out the ‘cancer’ of bad science, while leaving untouched any science that may yet be valid and useful. And in arguing against RAO, I am actually arguing for a more recent origin of humanity, though this is not my focus here and will not come through strongly in this article.
RAO makes a number of approximations, as do all theories by necessity. For this reason, almost any theory can be attacked for being ‘unrealistic’. The goal of this paper is to highlight the places where they make unrealistic assumptions in their favour, and, by pointing out how unrealistic these assumptions are, I hope to dispel some of the RAO mythology.
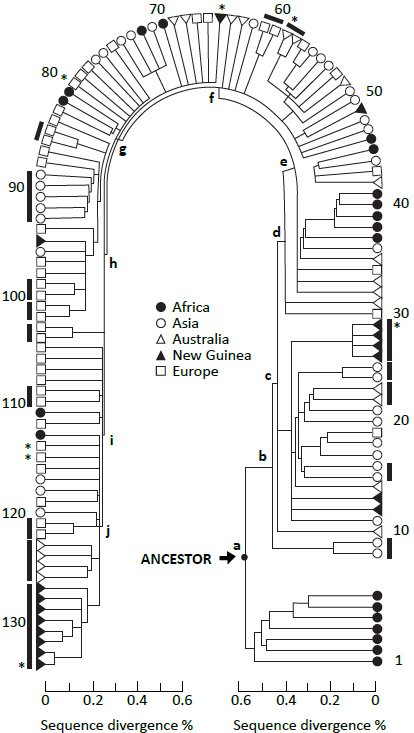
RAO does not support large-scale, deep-time evolution. There is nothing evolutionary about humans moving out of, or into, Africa at any time. Rather, the importance of the theory lies in the issue of dating and rooting the human genealogical tree. The following quote from the seminal RAO paper is going to be the focus of everything that follows: ‘We infer … that Africa is a likely source of the human mitochondrial gene pool. This inference comes from the observation that one of the two primary branches leads exclusively to African mtDNAs …, while the second primary branch also leads to African mtDNAs.’2 If we can dissect this, we will be far along the road to a better theory of human genetic history (figure 1).
The Standard Neutral Model of evolution
RAO is based on the Standard Neutral Model of evolution (SNM), itself being based on a long line of theoretical arguments, starting with J.B.S. Haldane’s writings in the 1950s. It is important to understand the development of this theory if we are to understand RAO.
Haldane (1957)3 was the first to discuss the concept known as the ‘cost of substitution’. According to Haldane, natural populations should not be able handle the number of deaths required by natural selection to drive positive evolution. Put simply, higher vertebrates do not have a high enough reproductive rate to support rapid rates of beneficial evolution. It takes too many deaths (the ‘cost’) to select for new mutations. Many creationists argue ‘Haldane’s Dilemma’ has not been sufficiently answered to date.4
Motoo Kimura took Haldane’s argument one step further by applying it to measured genetic differences between mammals. Following Haldane, he reasoned that if the genetic differences between two species were all due to positive selection, and if they evolved within the standard evolutionary timescale, then mammals would have needed an astronomically high reproductive rate to give natural selection enough fodder to drive evolutionary changes. For example, humans and chimpanzees have millions of genetic differences, but 3 million years would give us only about 100,000 human generations in which to fix these millions of differences. According to evolutionary calculations, natural selection would have had to remove many times more people than could possibly have been born during this time in order fix this many differences. To solve the dilemma, Kimura reasoned the majority of new mutations must be ‘neutral’ (this is the origin of the belief that most of the genome is composed of ‘junk DNA’, a term discussed elsewhere in this journal5). The rate of neutral evolution could be much faster than positive evolution, and would be limited only by the rate of DNA copying errors. Since natural selection will not act on neutral traits, which do not affect survival or reproduction, neutral evolution can proceed through random drift without any inherent cost of selection. Kimura saw Haldane’s Dilemma as a serious problem and listed it as his main reason for proposing the Neutral Theory of evolution.6 Haldane was right, Kimura asserted, but the preponderance of biological change must be neutral.
The Neutral Model has been expanded by many authors to become what we will call the Standard Neural Model (SNM). SNM was developed to cover complex historical patterns such as bottleneck events. It is the fundamental underpinning of RAO.
The following is a list of assumptions critical to the RAO model. Note how SNM is intrinsic to RAO theory (figure 2):
Constant population size. Although the Neutral Model is unaffected by population size, SNM was developed partly to model changes in population size (specifically one cycle of population bottleneck). RAO assumes a single human population with a single expansion event and no subsequent sub-population bottlenecks or other demographic differences.
Random mating (no population substructure, geographic or otherwise). Again, Neutral Theory is not contingent upon random mating, but RAO is. Population substructure prior to or after the Out of Africa Event might hide the true picture of human demographic history.
Neutral polymorphisms. There can be no selection acting on the alleles under question. RAO is based on the assumption that the entire mitochondrial genome is essentially neutral, or at least that negative mutations are efficiently eliminated. For detailed critique of this, see Sanford.7
An infinite-sites model of mutation. There cannot be multiple mutations at identical sites in different lineages and back mutations are not allowed. In some sense, this is a reasonable approximation, given the large size of the genome and the small number of generations involved (in the Creation model) or with the low rate of mutation (in evolutionary models). The approximation also simplifies the theoretical understanding and calculations. However, if this approximation turns out to be incorrect, RAO becomes harder to understand and the calculations behind it become less tenable.
Constant mutation/substitution rate among all subpopulations (the ‘molecular clock’). To illustrate how critical this is, I will quote Cann et al.:2 ‘A time scale can be affixed to the tree … by assuming that mtDNA sequence divergence accumulates at a constant rate’.

Constant effective generation time among all subpopulations (typically assumed to be 20 years for humans, although values between 20 and 30 have been used by various authors).
A human-chimp common ancestor some 3–6 million years ago. This is not an assumption of SNM, but is needed for calibrating the SNM bottleneck event. Essentially, by counting the number of differences between chimp and human mitochondria, and by then dividing this number by 3 to 6 million, one can get an estimate of the number of mutations that supposedly accumulate in the populations per year.
Tajima’s D statistic
Tajima’s D statistic8 is used to test SNM along a given stretch of DNA. It is basically a summary of the allele frequency spectrum. D values not significantly different from zero indicate the population meets all the criteria for SNM. Positive and negative values are due to an overabundance or dearth in the expected number of rare polymorphisms, respectively. Significant positive or negative values of D may indicate the presence of natural selection or historic changes in population size or multi-population introgression. This is an important metric for both SNM and RAO theory.
Historic changes in population size are expected to influence Tajima’s D statistic in that population growth should lead to excess low-frequency polymorphisms (negative D-values). This occurs because new mutations are carried along with the expansion and do not exit the population as easily through random drift. Alternatively, population bottlenecks should lead to a deficiency in low-frequency polymorphisms (positive D-values) because most low frequency alleles are eliminated through random selection.
Linkage disequilibrium
SNM needs to be put in the context of sexual reproduction. During gamete production, crossing over occurs between chromosome copies. This mixing causes randomization of the alleles (variations) along a stretch of DNA. However, because there are only one or two crossing over events per chromosome arm per generation, not all alleles are randomized each generation. And, the closer two alleles are, the less likely they are to be separated. Alleles in close proximity are said to be ‘linked’. This is also true of alleles separated by regions of infrequent crossing over. Geneticists use the term ‘linkage disequilibrium’ (LD) to describe the unequal association of certain alleles with certain other alleles. A set of alleles inherited together are referred to as a ‘haplotype’.
Population growth not only leads to negative D values (excess low frequency alleles), it also leads to less LD because crossing over randomizes more and more alleles each generation. Alternatively, population bottlenecks should lead to higher LD because during a bottleneck event a small number of people pass their large linkage blocks on to the entire population. It takes time for the haplotypes to be scrambled.
Expectations of SNM
The population parameters used in SNM calculations must be estimated, and this is no easy task. For humans, the mutation rate is based on current levels of genetic diversity, the assumption that the vast majority of that genetic diversity is neutral, and an assumed chimp-human ancestor 3–6 Ma. In other words, the calculation assumes large-scale evolution (i.e. macroevolution) is true. The effective population size (Ne) is also estimated from observed levels of diversity. But Ne is also dependent upon the generation time and the time to a common ancestor, two parameters about which little is known.9 Given an equal number of males and females and random mating, the ratio of hypothetical Ne for the autosomes, X chromosome, non-recombining portion of the Y chromosome (NRY), and mtDNA is 4:3:1:1, respectively. The reduced Ne is expected to produce more rapid differentiation among populations for the haploid loci than for the others.10This is one reason why RAO was initially based on mitochondrial sequences.
Since the calculation of the time to a most recent common ancestor (TMRCA) is directly proportional to Ne, TMRCAs for the autosomes and the X chromosome are expected to be 4 and 3 times greater, respectively, than for the two haploid loci.10 But this ignores the possibility that there was only one male lineage (Noah’s Y chromosome) and only three female lineages (the mitochondrial chromosomes of Noah’s three daughters-in-law) in the founding human population. The bottleneck event associated with the Flood would have created a strong signal that should still be evident today. Any model of human history that does not take this into account will come to incorrect conclusions if the Flood story is accurate.
SNM assumes that differences in LD among populations are due to differences in demographic history.11,12 This assumption depends, of course, on a constant rate of recombination, mutation and gene conversion in all subpopulations and a lack of ancient population structure (all of which affect LD). However, all it takes is one change in a DNA repair enzyme, one change in a gene that affects the rate of recombination, or one change in a gene that affects the process of gene conversion in one of the populations and the SNM results will diverge from reality. Thus, SNM assumes populations do not diverge in respect to certain genetic traits but are free to diverge in respect to others. This is perhaps the most critical point to understand.
According to SNM, low frequency alleles should be generally younger. As new alleles appear in the population, most of them will be lost through random drift. In fact, a new allele has a 1/Ne chance of eventually becoming fixed. In large stable populations, nearly all low frequency alleles are expected to be young. Younger alleles are also generally associated with longer haplotypes than high frequency alleles13 since it takes time to break down linkage blocks and shuffle new alleles in relation to older ones. Recombination is assumed to be neutral and random and to occur at a higher rate than mutation.10
By definition, the more linkage blocks a population has, the greater its expected ability to maintain polymorphism (linkage increases the variance of the numbers of polymorphic sites14). Higher levels of Ne also allow a population to maintain polymorphism (and a higher numbers of linkage blocks). The conclusion that African populations have maintained a larger long-term effective population size than non-African populations14 is based on the levels of polymorphism found in the populations, under the assumptions of SNM. But are the assumptions valid?
Violations of SNM
Violations of every assumption behind SNM have been detected and published in the evolutionary literature. Real-world populations do not conform to the constraints of SNM. Therefore, we must be careful when reading the conclusions of the many studies that have been based on this evolutionary theory.
SNM is a necessary simplification that allows for the study of a highly complex system. However, violations of SNM are expected at all levels: population structure, subpopulation bottlenecks, small-scale variations in recombination rates and multi-population admixture all increase estimates of LD9 and interfere with SNM calculations. Also, haplotype patterns can be disrupted by recurrent mutation, gene conversion, genome assembly errors and errors in genotyping.12 When deviations from the SNM occur (e.g. the conclusions of the majority of studies performed on the European population), the meaning of the estimated population parameters is unclear.14 In the real world, it is understood that generations overlap, that Ne fluctuates over time, that gene flow between populations changes over time and that population structure occurs within populations worldwide. Therefore, violations of SNM should be expected to occur frequently. One may rightly question the utility of a model that cannot be fit to real-world data.
Ne
One needs a good estimate of Ne in order to perform most SNM calculations, but Ne estimates are affected by several parameters, the effects of which must often be discounted in order for the model to run. Calculations of long-term Ne are disproportionately affected by low values (population contractions):10 therefore, the reported values should tend to be underestimates. Generation time also heavily influences Ne . In general, greater generation times are expected to result in proportionally lower Ne (and visa versa). Most LD studies assume generation times are equal among populations, something that cannot be historically proven. Cultural as well as genetic differences may lead to differences in generation times. Also, estimates of generation time from modern genealogical data are greater than the 20-year generation time commonly assumed in these studies (for example, 10 generations in my family tree = 300 years, or 30 years/generation). And, according to the biblical data, there should not be a constant generation time, because generation time decreased significantly immediately after the Flood.
Higher levels of Ne are expected to result in less genetic drift, slowing the divergence of populations. Human Ne has often been estimated to be about 10,000 people. Interestingly, due to their calculated inbreeding coefficient, Reich et al.11 estimated a historic human Ne of ‘50 individuals for 20 generations; 1,000 individuals for 400 generations; or any other combination with the same ratio.’ I would like to point out that 5 individuals for 2 generations fits their ratio and corresponds roughly with with biblical expectations. Is this evidence of the Flood (six people for one generation), or are these calculations so entrenched in evolutionary theory that they are not useful? Frisse et al.14 found disagreement between estimates of Ne for non-African populations based on LD and polymorphism data. This is another example of real population data in conflict with SNM assumptions. Which Ne should one use?
For unexplained reasons, the estimate of a historic human Ne of 10,000 individuals is much less than Ne estimates for the great apes.10 There is a huge amount of diversity among living chimpanzees:15 perhaps as much as three to four times as much diversity as within the entire human population.16 Does this evidence support evolution under an SNM scenario, or is the diversity not so much evidence of ancient Ne as much as it is evidence of a chimpanzee genome in rapid decline?
The calculation of Ne assumes deep time. In the biblical model, calculations of Ne are not done for there is no assumption of long ages. Rather, we say that there was a population bottleneck some 4,500 years ago (the Flood) where the world population was reduced to 3 founding couples (Noah’s sons and daughters-in-law).
Non-random crossing over events
Because we do not know much about the mechanism controlling crossing over events, nor the frequency in which they occur, it is often assumed that crossing over is more or less random. This allows for easier calculations of LD. However, the phenomenon is not entirely random. Recombination ‘hot spots’ have been known for years. It is believed that there is extensive fine-scale variation of recombination frequency within the human genome, and models that incorporate recombination hot spots are often better than ones that assume random recombination.17 We need to get a better understanding of recombination in order to better understand human genetic history. There is much room for improvement and much hope for the biblical model of human origins in this subject. The very presence of long, unmixed linkage blocks suggests a young genome, but we need more data.
Population structure
Random mating, or lack of population structure (specifically for sub-Saharan African population prior to the Out of Africa dispersion), is another key assumption behind the SNM. The effective rate of recombination is expected to be reduced in structured populations because haplotypes constrained within the various subpopulations will not have a chance to recombine as often as they would under panmixia.18 Importantly, failure to recognize population structure can lead to false positives when testing for constant Ne.1 When discussing the possibility of population structure in the presumed ancestral African population, Garrigan and Hammer10 worried that ancestral population structure could have had the effect of increasing ancestral Ne, thus throwing off their calculations. Behar et al.19 claimed that the early evolutionary history of man in Africa involved small and isolated tribes existing independently for thousands of years. This is a critical issue, for small and isolated populations experience inbreeding, rapid drift and the rapid accumulation of new mutations. The situation violates the fundamental assumptions behind the SNM while providing potentially excellent material to support the biblical model: for if several of the sub-Saharan African populations existed in such a condition after the Flood, this might go a long way in explaining why there is more genetic diversity among people of African descent.
The random mating model ignores reality, for population structure is a fact of human existence.20 Individuals have a tendency to choose mates from the same social groups21 and subpopulations in close proximity may be completely isolated from one another. In a study by Bulayeva et al.,22 they showed through genealogical analysis that in a certain village of 2,700 people in Daghestan, only 10 marriages had been consummated by the villagers with outside people over nine generations! And most of these marriages were with neighbouring villages. Bamshad et al.23 showed that the Hindu caste system has preserved a significant event in history—a huge invasion of India from the northwest. Men and women from upper castes are genetically more similar to Eastern Europeans, while those from lower castes are more similar to SE Asians. The religious system among a significant proportion of the Indian population has prevented free mixing for thousands of years! All this says that a realistic model of human demographic history would be overly complex,18 especially if it uses the wrong model of human history. A highly complex model may not be needed however (e.g. Liu et al. 200624), but there are always dangers involved in oversimplification.
Population admixture
Not only is the assumption of no population structure invalid (both world-wide and within subpopulations), but the situation is made more complicated by the mixing of once-separated populations. When previously separated populations come back together, heterozygosity increases. The mixing of haplotypes that arose separately causes an increase of calculated LD, even at unlinked sites.14 Admixture causes substantial variation in genetic ancestry among individuals in a population.1 Interestingly, the block characteristics of a mixed population should be most similar to the populations with the lowest LD.25 In other words, mixing can mask significant amounts of LD. For example, the fascinating admixture of African Bantu females and Jewish males (the ancestors of the Lemba tribes in SE Africa) has created a population with an unusually large number of long linkage blocks (long-distance LD).26
Gene conversion
While LD is known to decrease due to the effects of crossing over, another, less well-known process called ‘gene conversion’ may be at work as well. Essentially, gene conversion is a process by which a section of DNA can copy itself onto a highly similar section of DNA in close proximity. It has been studied extensively in yeast but relatively little is known about the process in mammals.9,14,24 It is expected that gene conversion acts over short distances, breaking down LD between closely-linked markers where crossing over is less likely.9,14 If gene conversion is allowed in a model, calculations of Ne based on the crossing over parameter get much smaller.14 Even though little is known about gene conversion, one recent study concluded that models using crossing over plus gene conversion fit the data better than models with crossing over alone.18 However, the possibility of recurrent mutations complicates this picture by theoretically inflating the apparent level of recombination and biasing gene conversion rates upwards.14 Furthermore, high levels of recurrent mutation can make the phylogenetic signal completely invisible.27
Due to gene conversion, the term ‘NRY’ (the non-recombining portion of the human Y chromosome) is technically a misnomer, for extensive gene conversion (a form of recombination) has been detected in the male-specific regions of the Y chromosome.28,29 It is expected that high levels of gene conversion will slow divergence rates because they systematically eliminate mutational events.
Equivalent mutation rates and active selection
One of the central assumptions behind SNM is that equivalent genetic sequences in diverse populations evolve in a clock-like manner. This can only happen if selection is not acting upon the genetic sequences under question and if mutation rates are equal among all populations. However, there is evidence that a molecular clock is not operating within related clades of African mitochondrial haplogroup L2. Two of the four clades studied by Torroni et al.30 were ‘disproportionately derived’ and they concluded that their results were ‘not consistent with a simple model of neutral evolution with a uniform molecular clock.’ Howell et al.31 indicated that there may be clock violations for all African L mitochondrial haplogroups and that there were differences in clock rates between coding and control regions. Friedlaender et al.32 went so far as to say that the variable mutation rates among mtDNA clades bring the utility of coalescent statistics and associated age estimates into question.
These are key findings, for the deep-rooting branches of the sub-Saharan lineages are foundational to RAO theory. If no molecular clock is operating among them, there is no way to time the African diaspora. And the only basis for claiming these lineages are ancestral is that they are more divergent from the rest of the world population … but how they became so divergent would then be an open question.
Other studies have concluded that deviations from SNM in Europe and Asia may be due to natural selection. Also, natural selection may not be equal among all clades, especially since clades are not distributed equally across all environmental regions.33,34
Cultural factors may also mimic selection. Genghis Khan is ancestor to a surprising number of people in Central and Eastern Asia and is ancestor to perhaps 0.5% (1 out of 200!) of the world’s population.35 If we did not know about the existence of Genghis Khan from historical sources, how would this missing information affect our conclusions about the distribution of Asian Y chromosomes? And if non-random events like this can have such a profound effect on genetic diversity patterns, how could we trust molecular ‘clocks’ that depend so heavily on random mutation and random mating?
Conclusions
The studies referenced in this paper highlight the tenuous nature of the Recent African Origins theory. RAO relies deeply on the Standard Neutral Model of evolution, but every assumption behind SNM and, therefore, RAO has been openly questioned in the evolutionary literature. If the African sequences are not evolving in a clock-like manner, the Recent African Origins theory must be seriously reworked. The same would be true if selection is operating on the non-African sequences. If real-world populations violate the central assumptions of SNM, the conclusions of studies that assume SNM are not and cannot be the final word on the subject.
We can conclude that the assumptions behind SNM are not completely realistic. The real situation is much more complicated than the simplifying assumptions allow (figure 3) and several of these assumptions are either biased in favour of the conclusion or contrary to the data. Crossing over is not random. Population structure exists at all scales. Population admixture (especially if it occurred in the distant past) and gene conversion overly complicate the models. The presence of natural selection among human mitochondria removes the ‘neutral’ part of the SNM as far as RAO’s Mitochondrial Eve is concerned.
Finally, RAO and SNM are based on the belief that evolution can occur, but that it cannot affect the things that control the speed of evolution. There is no room for differences in mutation rates30,31 among populations caused by environmental stress,33,34,36 demographics (life-history patterns caused by cultural factors), or mutations in DNA polymerase and the DNA copying, proofreading or correcting mechanisms.37,38
It has now been shown that the most popular evolutionary model of recent human evolution is unsatisfactory. But with what shall we replace it? The biblical model for human genetic history is still in its infancy. I hope this short article will spark creative thinking in other creation scientists, who will take up the torch by attacking evolutionary theory at its roots: but also, that they will succeed in introducing new ideas to the community at large. There is much work to be done. I have only sketched an outline and I have only hinted at several lines of thought that might be quite productive to creationist research.
Acknowledgments
The writing of this paper was heavily influenced and partially supported by J. Sanford. I would like to thank one of the reviewers in particular for an excellent critique; the manuscript was significantly improved as a result.
References and notes
- Rosenberg, N.A. et al., Genetic Structure of Human Populations, Science 298:2381–2385, 2002. Return to text.
- Cann, R.L., Stoneking, M. and Wilson, A.C.. Mitochondrial DNA and human evolution, Nature 325:31–36, 1987. Return to text.
- Haldane, J.B.S., The cost of natural selection, J. Genetics 55:511–524, 1957. Return to text.
- ReMine, W.J., Cost theory and the cost of substitution—a clarification, J. Creation 19(1):113–125, 2005. Return to text.
- Woodmorappe, J., Junk DNA indicted, J. Creation 18(1):27–33, 2004. Return to text.
- Kimura, M., Evolution rate at the molecular level, Nature 217:624–626, 1968. Return to text.
- Sanford, J., Genetic Entropy and the Mystery of the Genome, FMS Publications, Waterloo, NY, 2008. Return to text.
- Tajima, F., Statistical-method for testing the neutral mutation hypothesis by DNA polymorphism, Genetics 123:585–595, 1989. Return to text.
- Ptak, S.E., Voelpel, K. and Przeworski, M., Insights into recombination from patterns of linkage disequilibrium in humans, Genetics 167:387–397, 2004. Return to text.
- Garrigan, D. and Hammer, M.F., Reconstructing human origins in the genomic era, Nature Reviews Genetics 7:669–680, 2006. Return to text.
- Reich, D.E. et al., Linkage disequilibrium in the human genome, Nature 411:199–204, 2001. Return to text.
- Gabriel, S.B. et al., The structure of haplotype blocks in the human genome, Science 296:2225–2229, 2002. Return to text.
- Voight, B.F. et al., A map of recent positive selection in the human genome, PLoS Biology 4(3):446–458, 2006. Return to text.
- Frisse, L. et al., Gene conversion and different population histories may explain the contrast between polymorphism and linkage disequilibrium levels, Am. J. Human Genetics 69:831–843, 2001. Return to text.
- Becquet, C. et al., Genetic structure of chimpanzee populations, PLoS Genetics 3(4):617–626, 2007. See also sciencedaily.com. Return to text.
- Kaessmann, H., Wiebe, V. and Pääbo, S., Extensive nuclear DNA sequence diversity among chimpanzees, Science 286:1159–1162, 1999. See also sciencedaily.com. Return to text.
- Wall, J.D. and Pritchard, J.K., Assessing the performance of the haplotype block model of linkage disequilibrium, Am. J. Human Genetics 73:502–515, 2003. Return to text.
- Przeworski, M. and Wall, J.D., Why is there so little intragenic linkage disequilibrium in humans? Genetic Research, Cambridge 77:143–151, 2001. Return to text.
- Behar, D.M. et al., and The Genographic Consortium, The dawn of human matrilineal diversity, Am. J. Human Genetics 82:1130–1140, 2008. Return to text.
- Cavalli-Sforza, L.L., Menozzi, P. and Piazza, A., The History and Geography of Human Genes, Princeton University Press, Princeton, NJ, 1994. Return to text.
- Rohde, D.L.T., Olson, S. and Chang, J.T., Modelling the recent common ancestry of all living humans, Nature 431:562–566, 2004. Return to text.
- Bulayeva, K.B. et al., Ethnogenomic diversity of Caucasus, Daghestan, Am. J. Human Biology 18:610–620, 2006. Return to text.
- Bamshad, M. et al., Genetic evidence on the origins of Indian caste populations, Genome Research 11:994–1004, 2001. Return to text.
- Liu, H. et al., A geographically explicit genetic model of worldwide human-settlement history, Am. J. Human Genetics 79:230–237, 2006. Return to text.
- Wall, J.D. and Pritchard, J.K., Haplotype blocks and linkage disequilibrium in the human genome, Nature Reviews Genetics 4:587–597, 2003. Return to text.
- Wilson, J.F. and Goldstein, D.B., Consistent long-range linkage disequilibrium generated by admixture in a Bantu-Semitic hybrid population, Am. J. Human Genetics 67:926–935, 2000. Return to text.
- Torroni, A. et al., Harvesting the fruit of the human mtDNA tree, TRENDS in Genetics 22(6):339–345, 2006. Return to text.
- Rozen, S. et al., Abundant gene conversion between arms of palindromes in human and ape Y chromosomes, Nature 423:873–876, 2003. Return to text.
- Skaletsky, H. et al., The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes, Nature 423:825–838, 2003. Return to text.
- Torroni, A. et al., Do the four clades of the mtDNA haplogroup L2 evolve at different rates? Am. J. of Human Genetics 69:1348–1356, 2001. Return to text.
- Howell, N. et al., African haplogroup L mtDNA sequences show violations of clock-like evolution, Molecular Biology and Evolution 21(10):1843–1854, 2004. Return to text.
- Friedlaender, J. et al., Expanding Southwest Pacific mitochondrial haplogroups P and Q, Molecular Biology and Evolution 22(6):1506–1517, 2005. Return to text.
- Mishmar, D. et al., Natural selection shaped regional mtDNA variation in humans, Proceedings of the National Academy of Sciences (USA) 100(1):171–176, 2003. Return to text.
- Moilanen, J.S., Finnilä, S. and Majamaa, K., Lineage-specificselection in human mtDNA: lack of polymorphisms in a segment of MTND5 gene in haplogroup J, Molecular Biology and Evolution 20(12):2132–2142, 2003. Return to text.
- Zerjal, T. et al., The genetic legacy of the Mongols, American Journal of Human Genetics 72:717–721, 2003. Return to text.
- Ames, B., Low micronutrient intake may accelerate the degenerative diseases of aging through allocation of scarce micronutrients by triage, Proceedings of the National Academy of Science (USA) 103(47):17589–17594, 2006. Return to text.
- Copeland, W.C. et al., Mutations in DNA polymerase gamma cause error prone DNA synthesis in human mitochondrial disorders, Acta Biochemica Polonica 50(1):155–167, 2003. Return to text.
- Lee, H.R. and Johnson, K.A., Fidelity of the Human Mitochondrial DNA Polymerase, J. Biological Chemistry 281(47):36236–36240, 2006. Return to text.
- Carter, R. Mitochondrial diversity within modern human populations, Nucleic Acids Research 35(9):3039–3045, 2007. Return to text.








Readers’ comments
Comments are automatically closed 14 days after publication.