

Journal of Creation 25(2):111–117, August 2011
Browse our latest digital issue Subscribe
The chromosome 2 fusion model of human evolution—part 2: re-analysis of the genomic data
A major argument for human evolution from a shared common ancestor with the great apes, particularly chimpanzees, is the ‘chromosome 2 fusion model’. This molecular model involves the hypothetical fusion of two small acrocentric chimpanzee-like chromosomes (2A and 2B) at some ancient point in the human evolutionary lineage. Our analysis of the available genomic data shows that the sequence features encompassing the purported chromosome 2 fusion site are too ambiguous to accurately infer a fusion event. The data actually suggest that the core ~800 bp region containing the fusion site is not a unique cryptic and degenerate head-to-head fusion of telomeres, but a distinct motif that is represented throughout the human genome with no orthologous counterpart in the chimpanzee genome on either chromosome 2A or 2B. The DNA sequence evidence for a purported inactivated cryptic centromere site on chromosome 2, supposedly composed of centromeric alphoid repeats, is even more ambiguous and untenable than the case for a fusion site. The alphoid sequences in this region are quite variable and do not cluster with known functional human centromeric sequences. In addition, no ortholog for a cryptic centromere homologous to the alphoid sequence at human chromosome 2 exists on chimpanzee chromosomes 2A and 2B.

One of the most cited DNA-based arguments for human evolution is the hypothetical head-to-head fusion of two small ape-like chromosomes to form human chromosome 2.1 The corresponding chromosomes supposedly represented in the great apes are 2A and 2B in the chimpanzee genome. A majority of the research that undergirds this model utilized indirect methods of DNA analysis. These data were derived from DNA probe hybridization, chromosomal banding (staining), and limited DNA sequencing techniques that were available prior to the advent of high-throughput DNA sequencing technology.1,2
Chromosome staining and hybridization techniques do not provide detailed DNA sequence information, but rather indicate putative areas of homology. Chromosome staining used to achieve visible banding markers yields information related to GC base content, repeat content, CpG island density, and degree of condensation over large areas rather than specific sequence homology.3,4 Probe (DNA) hybridization is a more direct and accurate method for detecting DNA homology, but is subject to lab protocol variability and does not provide actual DNA sequences. Early DNA sequencing projects were largely limited to small, isolated regions of eukaryote genomes, a scenario that changed with the introduction of large-insert DNA cloning (bacterial artificial chromosomes; BACs) and BAC contig-based physical mapping strategies.
The advent of high-throughput DNA sequencing and its accompanying technologies has largely replaced these earlier technologies for comparing both chromosomes and genomes. The first working draft of the human genome generated in both the public and private sectors was available in 2001 and a more complete draft of the public human genome sequence became available in 2003.5–7 The chimpanzee genome project also received funding, and a 5-fold redundant shotgun sequence coverage was published in 2005.8 Another 1.5-fold coverage was completed after this along with the construction of a BAC contig-based physical map for chimpanzee.9
While the chromosome 2 fusion model has been routinely discussed in reviews of human evolution, very little new supporting genomic data, although readily available for analysis, has been forthcoming. For the purpose of propagating the dogma surrounding human evolution, several science authors have recently published novice-level science books promoting the hypothetical chromosome 2 model.10,11 This so-called factual data is routinely used as one of the leading arguments for human evolution from a shared common ancestor with apes.
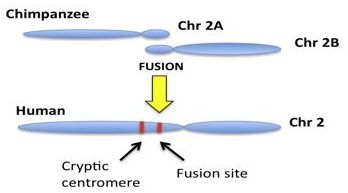
The general model involves the hypothetical fusion of two small, acrocentric,12 ape-like precursor chromosomes thought to have fused end-to-end, forming the single large human chromosome 2, as illustrated in figure 1. From a DNA sequence perspective, it is claimed that human chromosome 2 contains two key regions in its landscape. The first region of interest is thought to depict the actual head-to-head fusion of telomeres. Telomeres are end-cap DNA repeat motifs (TTAGGG)n located at the termini of linear mammalian chromosomes, recently reviewed by Tomkins and Bergman.13 The second key region supposedly represents a cryptic non-functional centromere that was inactivated following the fusion event. For each chromosome, a single functional centromere is required for proper stability and function because a dual centromere situation created by such a fusion would cause cellular instability and destruction. Although there are no well-defined mechanisms for inactivating human centromeres, it is believed that one of the two resulting centromeres was somehow silenced as a result of fusion. The chromosome 2 fusion is thought to account for the fact that humans have only 46 (2N) chromosomes and the great apes, including chimpanzee have 48 (2N). Modern humans supposedly evolved from a shared common ancestor with a diploid genome of 48 chromosomes, requiring a fusion event.
Examining the genomic evidence for fusion
Of the two genomic regions that are claimed to support the fusion model, the primary evidence is the purported fusion site. This site is located in a region close to the present functional centromere on the long arm of human chromosome 2. This particular area containing the ‘fusion region’ is often called 2qfus or 2chr2fus and occupies the genomic area between 2q13 and 2q14.1.14 The two small chimpanzee chromosomes that supposedly contributed to the fusion event are currently identified as 2A and 2B.
The human 2qfus region has been sequenced and annotated for telomeric repeats, a variety of important functional genes, processed pseudogenes, and various open reading frames (ORFs). A fairly thorough and complete 614 kb (614,000 bases) annotated genomic landscape was constructed that encompasses the fusion site and was published by a lab in several related reports shortly after the initial first working draft of the human genome project.15,16 The primary substrate for the effort relied on the assembled sequence from five overlapping, large-insert DNA clones (bacterial artificial chromosomes; BACs). As a result of this effort, a 177 kb region of contiguous sequence directly surrounding the 2qfus site corresponding to BAC clone RP11-395L14 (accession number AL078621) is available for public access and download. For the purpose of clarifying claims related to the fusion site, we subjected the complete BAC sequence of RP11-395L14 to a variety of telomere motif analyses (see Materials and Methods).
Fusion site DNA sequence analysis
Our DNA sequence analysis confirmed conclusions reached by Fan et al. The putative fusion site is ‘highly degenerate’ and a vague shadow of what should be present given the model proposed.15 One of the major problems with the fusion model is that, within the 20- to 30-kb window of DNA sequence surrounding the hypothetical fusion site, there is a glaring paucity of telomeric repeats, and those that are present are mostly independent monomers, not tandem repeats. In fact, many of the motifs in the 30-kb region surrounding the putative 2qfus site are not only isolated monomers, but are separated by up to several thousand bases of DNA.
Even while completely disregarding a consensus 6-base reading frame when iterating through the repeats, for the left (plus strand) side of the fusion site, there are only 34 intact TTAGGG motifs (table 1). This analysis uses a generous allowance of 92,690 bases to the left of the fusion site where the first TTAGGG repeat is found on BAC RP11-395L14, well beyond the size of any normal human telomere. Based on the predicted model, thousands of intact TTAGGG motifs in tandem should exist. This is true even if allowing for an extremely high rate of degeneracy, which is an unreasonable expectation because meiotic recombination is suppressed in pericentric DNA due to its close proximity to the centromere. Recombination, the most likely theoretical source of sequence shuffling leading to the fusion site degeneration would therefore be less of consideration. Also, based on the predicted model, little, if any TTAGGG motifs should exist on the plus strand to the right of the fusion site. However, 18 intact TTAGGG motifs are found on the right of the fusion site; 35% of the total number of TTAGGG motifs located within a generous 156,911 base window surrounding the fusion site.
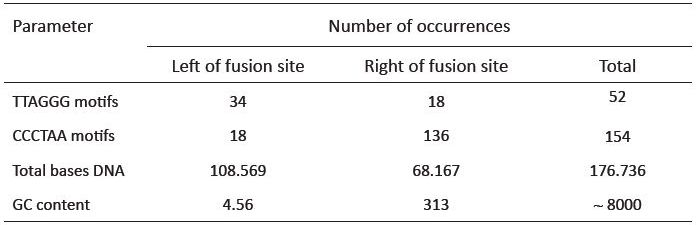
The reverse complement telomere sequence (CCCTAA) should be present in near-perfect tandem to the right of the fusion site. Like the TTAGGG motif, one would expect approximately 1667 to 2500 CCCTAA motifs if an end-to-end fusion occurred. However, only 136 intact motifs exist to the right of the fusion site, with the last CCCTAA on the BAC clone terminating at 64,221 bases to the right of the fusion (table 1). Again, this very generous stretch of sequence is much longer than a normal human telomere, and contains a paucity of motifs. In similar fashion to the TTAGGG forward motif, the CCCTAA motif was also located on both sides of the fusion site. Our analysis located a total of 18 occurrences of the CCCTAA motif (12% of the total) scattered throughout the opposite side of the fusion site, where it would not be expected to be found. In other words, both the forward and reverse complement of the telomere motif populate both sides of the fusion site. As a side note, the GC content of the 177 kb region encompassing the putative fusion site is significantly higher (45%) than the average (40%) for chromosome 2 (table 2).

A complete scan of the 237+ million bases of the assembled euchromatic sequence of chromosome 2 using the Skittle Genome Viewer software package showed that the entire landscape, from end to end, is populated with TTAGGG and CCCTAA motifs. Small, isolated dense clusters of telomere motifs occurred in at least 5 internal locations (data not shown). A complete iteration of the entire plus strand sequence of chromosome 2 (Per script written by Tomkins) indicated a total number of ‘TTAGGG’ and ‘CCCTAA’ occurences at 45,450 and 45,770, respectively (table 2). These numbers are roughly equal, indicating that both the forward and reverse orientation of the telomere motif occurs quite frequently at internal sites across the length of chromosome 2. These numbers indicate that a total of at least 547,320 internal bases on chromosome 2 are composed of widely distributed intact telomere motifs.
An important attribute associated with these internal telomere motifs is that they are largely monomeric. Of the 52 intact TTAGGG motifs on both sides of the fusion site, only three tandem occurrences were found, with the rest existing as independent monomers. Of the 154 intact CCCTAA motifs on both sides of the fusion site, eighteen tandem motifs were found, with the rest appearing as independent monomers. Although the density of motifs and dimeric repeats increases somewhat within the immediate vicinity of the putative fusion region, their positions in the reading frame from one 6-bp telomeric repeat to the next are erratic (not in frame).
Because of the extreme paucity of telomeric repeats, their largely monomeric condition, and their ubiquitous presence on both sides of the supposed fusion site, there exists little data to indicate that they may have once formed 10- to 15-kb stretches of perfect, tandem 6-base repeats. The 2qfus sequence is clearly degenerate beyond the point of indicating that intact telomeres once existed. Given the location in a region of suppressed pericentric recombination, one would expect a considerably higher amount of telomere sequence preservation if the model was tenable.
In attempting to correlate rates of evolutionary change with the extreme degeneracy observed in the putative fusion region, one research group concluded that “the head-to-head repeat arrays at the RP11-395L14 fusion site have significantly degenerated from the near perfect (TTAGGG)n arrays found in telomeres.”15 This caused them to raise the question, “Why are the arrays at the fusion site so degenerate if the fusion occurred within the telomeric repeat arrays less than ~6 Mya?”15

A more valid explanation for the telomere-like features present at the putative fusion site is that they may represent some form of a distinct genomic motif. To test this idea, a 798-bp fragment (figure 2) encompassing the fusion site and the region where the telomeric motifs are more densely populated was used as a query subject in a BLAT17 search on the most recent build of the human genome (v 37.1; www.genome.usc.edu) with masking disabled. The results revealed a total of 159 significantly placed hits throughout the genome on human chromosomes 1–11, 15, 18–20, X and Y. The homologous regions for these hits included areas near telomeres, pericentric areas, and a wide variety of internal euchromatic sites. Identity values ranged from 80.5 to 100%, supporting the conclusion that the telomere fusion site core sequence is not unique to its pericentric location on chromosome 2, and instead represents a sequence feature (motif) scattered throughout the human genome.

To verify the BLAT results and to identify homologous sites in the chimpanzee genome, the BLASTN algorithm was used (with no masking or gap extension) for comparisons between the 798-bp core 2qfus sequence and the most recent builds of the human (v 37.1) and chimp (v 2.1) genomes maintained at NCBI (www.ncbi.nlm.nih.gov/). Although the BLASTN query against the human genome was more data intensive than the index-based BLAT search, the results produced a total of 85 significantly placed hits on all human chromosomes except chromosomes 13, 16 and 17 (1–12, 14, 15, 18–22, X and Y). While the number of hits was reduced, compared to BLAT, more chromosomes with homologous sites were identified with the BLASTN search because of the more direct nature of the algorithm (figure 3). Interestingly, human chromosomes 2, 16, 21 and 22 were peppered with the ‘fusion site’ sequence over the length of their entire euchromatic landscape (figure 3).
When the 798-bp core fusion sequence was BLASTN queried against the chimpanzee genome, the significantly placed hit count was reduced to 19, only 22% of the amount observed in the human genome. This is a startling find in light of the wide-spread claims that the human and chimpanzee genomes contain DNA sequence that is supposedly 96 to 98% similar, a claim perhaps related to the fact that the human genome was used as a scaffold to build the chimpanzee genome.8 In addition, the human-chimp hit locations did not show strong synteny, as only 13 of the 19 hits (68%) shared visually similar locations in the genome (on chimpanzee chromosomes 1, 2B, 8, 9, 12, 14, 15, 18, 20 and 22).
The most startling outcome of this analysis is that the fusion site did not align with chimp chromosome 2A, one of the supposed pre-fusion precursors. Furthermore, the alignment at two locations on chromosome 2B, an internal euchromatic site and the telomere region of its long arm, did not match predicted fusion-based locations based on the fusion model. If the fusion model was credible, this should have produced an alignment with the telomeric region on chimpanzee 2B on the short arm.
There is, therefore, no real evidence for DNA homology between human and chimpanzee for the 798-bp core fusion sequence. The alignment data also severely calls into question claims of high overall sequence similarity of 96 to 98% between the genomes. Our results are indirectly supported by the exceptionally high levels of dissimilarity observed in a recent study of a section of the Y chromosome landscape between human and chimpanzee.
Examining DNA sequence for a cryptic centromere
Following the supposed head-to-head telomere-based fusion of two smaller chromosomes, two centromeres would have had to exist in the newly formed chimeric chromosome, one from each of the two fused chromosomes. According to the evolutionary model, sequence degeneration plus selection would continue until the second centromere was completely non-functional. The DNA evidence in question is based on the fact that human, great-ape, and other mammalian centromeres are composed of a highly variable class of DNA sequence that is repeated over and over called alpha-satellite or alphoid DNA.18 Alphoid DNA, although found in centromeric areas, is not unique to centromeres and is even highly variable between homologous regions throughout the same mammalian genome.18
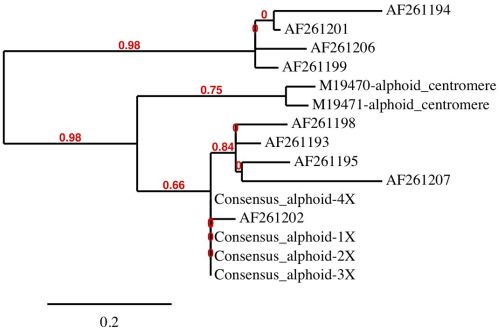
The basic human alphoid monomer is a 171-base motif represented by a patented synthetic consensus sequence in Genbank (Acc. # CS444613). There also exists two small sequenced clones representing alphoid repeats with proven cellular centromere function.19 Nine different alphoid fragments in the cryptic centromere site associated with the purported chromosome 2 fusion event were also sequenced and submitted to GenBank by an Italian laboratory (see figure 4 for accession numbers). In total, we downloaded and analyzed all 12 of these sequences for similarity to each other and individually for genome-wide homology.
Using the BLAT tool on the most recent version (v 3.7) of the human genome assembly, the nine italian lab alphoid sequences elicited the strongest hits at the chromosome 2 putative cryptic centromere site for all accessions. This confirmed that they were cloned from this region of the genome. The consensus 171-bp alphoid sequence aligned at the cryptic centromere site with 90.6% identity, supporting the conclusion that the site contains alphoid-like sequences.
However, the concern is not if this location contains alphoid sequences that are known to be ubiquitous in the human genome, but how similar these sequences are to each other and to known functional centromeric alphoid repeats. Alphoid sequences located at centromeres form long series of repeat patterns that are very homogeneous in their repetitive structure, producing distinctive higher-order patterns. Alphoid regions that are non-centromeric are more diverse in their monomer content and form higher order patterns with different characteristics compared to centromeres.20 At present, there are five known supraclasses of human alphoid monomers that combine in various combinations.21 There is also evidence from research in progress that alphoid monomer classes themselves can be broken down further into specific subfragments that may be present in the genome by themselves or as a sub-fragment in an alphoid repeat region (Tomkins, unpublished data).
In a human alphoid multiple-sequence alignment analysis, we combined the two functional centromeric alphoid sequences with the set of nine Italian alphoid sequences along with the consensus 171-base alphoid sequence in our data set (figure 4). We also created tandem repeats of the consensus 171-base alphoid sequence representing repeats of 2X to 4X in length as individual sequences. Alignments were conducted using the MUSCLE software package22 then refined using the Gblocks program.23
The human alphoid alignments clearly revealed dissimilarity between alphoid sequences and distinct patterns of clustering. Patterns of similarity were computationally evaluated using PhyML24 with tree rendering performed by TreeDyn (figure 4).25 Four major groups were distinguished by the PhyML analysis with the functional centromere sequences clustering by themselves and not with the alphoid sequences located at the purported cryptic centromere site on chromosome 2. The sequences at the cryptic centromere site are clearly a diverse mixture of alphoid monomers, forming three separate groups and not distinctly representative of functional centromeric DNA. In a structural comparison of both the functional centromere and cryptic centromere sites on chromosome 2 with the genome visualization tool, Skittle,26 the putative cryptic centromere site was considerably more sequence-diverse and structurally unordered compared to the functional centromere on chromosome 2 (data not shown). The complex higher-order architecture of this Alphoid-diverse site is clearly unique and not characteristic of a silenced degenerate centromere.
Multiple reports involving both hybridization and sequence-based research of alphoid/centromere similarity between humans and apes have found virtually no apparent evolutionary homology, except for moderate similarity on the X-chromosome centromere.20,27 Baldini et al. found that the “highest sequence similarity between human and great ape alphoid sequences is 91%, much lower than the expected similarity for selectively neutral sequences.”28 Alphoid regions, in contrast to many classes of DNA sequences, are not well-conserved among taxa and even show high levels of diversity between chromosomes in the same genome.18 When the human alphoid sequences in our data set were queried against the chimpanzee genome using both BLAT and BLASTN, we were unable to obtain a single significant hit, verifying the extreme dissimilarity observed in alphoid motifs between taxa. These data corresponded well with several decades of previous research by multiple labs, discussed above.
Summarized findings
- The reputed fusion site is located in a peri-centric region with suppressed recombination and should exhibit a reasonable degree of tandem telomere motif conservation. Instead, the region is highly degenerate—a notable feature reported by a previous investigation.
- In a 30 kb region surrounding the fusion site, there exists a paucity of intact telomere motifs (forward and reverse) and very few of them are in tandem or in frame.
- Telomere motifs, both forward and reverse (TTAGGG and CCTAAA), populate both sides of the purported fusion site. Forward motifs should only be found on the left side of the fusion site and reverse motifs on the right side
- The 798-base core fusion-site sequence is not unique to the purported fusion site, but found throughout the genome with 80% or greater identity internally on nearly every chromosome; indicating that it is some type of ubiquitous higher-order repeat.
- No evidence of synteny with chimp for the purported fusion site was found. The 798-base core fusion-site sequence does not align to its predicted orthologous telomeric regions in the chimp genome on chromsomes 2A and 2B.
- Queries against the chimp genome with the human alphoid sequences found at the purported cryptic centromere site on human 2qfus produced no homologous hits using two different algorithms (BLAT and BLASTN).
- Alphoid sequences at the putative cryptic centromere site are diverse, form three separate sub-groups in alignment analyses, and do not cluster with known functional human centromeric alphoid elements.
Materials and Methods
DNA sequences described in this paper were downloaded from the National Center for Biotechnology (NCBI) web site in FASTA format text files.29 Results from online BLAT (Blast-Like Alignment Tool)17 searches were downloaded from the Genome Browser at the UCSC Genome Bioinformatics web site (genome.ucsc.edu/) as plain text files and parsed using a POSIX shell script written by J.P. Tomkins. Analyses for telomere motif occurrence and GC content were performed using a Perl script written by J.P. Tomkins. Bioinformatic scripts developed and utilized in this study may be requested by contacting author Tomkins at jtomkins@icr.org. Figures depicting genome-view BLASTN (nucleotide BLAST) alignments were obtained using online software available at NCBI. For alphoid sequence alignments, the MUSCLE (Multiple Sequence Comparison by Log-Expectation)22 program (v 3.7; www.ebi.ac.uk/Tools/muscle/index.html) followed by curation with Gblocks (v 0.91b; molevol.cmima.csic.es/castresana/Gblocks.html)23 was used to evaluate alignments and select conserved blocks for analysis with PhyML (v 3.0; atgc.lirmm.fr/phyml/).24 Tree data from PhyML was rendered with TreeDyn (v 198; www.treedyn.org/).25 Sequence visualization of repeats and motif patterns were performed using the genome viewer software program Skittle26 and the entire consensus sequence of human chromosome 2 downloaded as a compressed fasta file from NCBI.
Acknowledgments
We wish to thank John Sanford, Robert Carter, Clifford Lillo, and Mary Ann Stuart for their reviews of an earlier draft of this manuscript, and Josiah Seaman and John Sanford for their guidance in the usage of the ‘Skittle’ genome viewer software.
References
- Yunis, J.J. and Prakash, O., The origin of man: a chromosomal pictorial legacy, Science 215:1525–1530, 1982. Return to text.
- Ijdo, J.W. et al., Origin of human chromosome 2: an ancestral telomere-telomere fusion, Proc. Natl. Acad. Sci. 88:9051–9055, 1991. Return to text.
- Furey, T.S and Haussler, D., Integration of the cytogenetic map with the draft human genome sequence, Human Mol. Genet. 12:1037–1044, 2003. Return to text.
- Niimura, Y. and Gojobori, T., In silico chromosome staining: reconstruction of giemsa bands from the whole human genome sequence, Proc. Natl. Acad. Sci. 99:797–802, 2002. Return to text.
- International Human Genome Sequencing Consortium, Initial sequencing and analysis of the human genome, Nature 409:861–920, 2001. Return to text.
- Venter, J.C. et al., The sequence of the human genome, Science 291:304–1351, 2001. Return to text.
- International Human Genome Sequencing Consortium, Finishing the euchromatic sequence of the human genome, Nature 431:931–945, 2004. Return to text.
- The Chimpanzee Sequencing and Analysis Consortium, Initial sequence of the chimpanzee genome and comparison with the human genome, Nature 437:69–87, 2005. Return to text.
- Warren, R.L., Physical map assisted whole-genome shotgun assemblies, Genome Res. 16:768–775, 2010. Return to text.
- Fairbanks, D.J., Relics of Eden, Prometheus Books, Amherst, NY, 2007. Return to text.
- Miller, K.R., Only a Theory: Evolution and the Battle for America’s Soul, Viking, New York, 2008. Return to text.
- Acrocentric chromosome has one arm that is considerably shorter than the other arm. The ‘acro’ in acrocentric refers to the Greek word for ‘peak’. The human genome contains five acrocentric chromosomes: 13, 14, 15, 21 and 22. Return to text.
- Tomkins, J. and Bergman, J., Telomeres: implications for aging and evidence for intelligent design, J. Creation 25(1):86–97, 2011. Return to text.
- This system uses p to represent the small arm of the chromosome and q to represent the long arm. The numbers refer to chromosome bands—sections of the chromosome that can be visualized by stains and dyes or other techniques. Thus, 2q13 refers to section (band) 13 on the long arm of chromosome 2. Return to text.
- Fan, Y. et al., Genomic structure and evolution of the ancestral chromosome fusion site in 2q13-2q14.1 and Paralogous Regions on Other Human Chromosomes, Genome Res. 12:1651–1662, 2002. Return to text.
- Fan, Y. et al., Gene content and function of the ancestral chromosome fusion site in human chromosome 2q13-2q14.1 and paralogous regions, Genome Res. 12:1663–1672, 2002. Return to text.
- Kent, W.J., The BLAST-Like Alignment Tool, Genome Res. 12:656–664, 2002. Return to text.
- Alkan, C. et al., Genome-wide characterization of centromeric satellites from multiple mammalian genomes, Genome Research 21:137–145, 2011. Return to text.
- Heartlein, M.W. et al., Chromosome instability associated with human alphoid DNA transfected into the chinese hamster genome, Molec Cell. Biol. 8:3611–3618, 1988. Return to text.
- Alkan C. et al., Organization and evolution of primate centromeric DNA from whole-genome shotgun sequence data, PLoS Comput Biol 3:1807–1818, 2007. Return to text.
- Roizes G., Human centromeric alphoid domains are periodically homogenized so that they vary substantially between homologues. Mechanism and implications for centromere functioning, Nucleic Acids Res 34:1912–1924, 2006. Return to text.
- Edgar, C.E., MUSCLE: multiple sequence alignment with high accuracy and high throughput, Nucleic Acids Res. 32:1792–1797, 2004. Return to text.
- Talavera, G. and Castresana, J., Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments, Systematic Biol. 56:564–577, 2007. Return to text.
- Guindon, S. et al., PHYML Online—a web server for fast maximum likelihood-based phylogenetic inference, Nucleic Acids Res. 33:W557–W559, 2004. Return to text.
- Chevenet, F. et al., TreeDyn: towards dynamic graphics and annotations for analyses of trees, BMC Bioinformatics 7:439, 2006. Return to text.
- Seaman, J.D. and Sanford, J.C., Skittle: A 2-dimensional genome visualization tool, BMC Bioinformatics 10:452, 2009. Return to text.
- Archidiacono, N. et al., Comparative mapping of human alphoid sequences in great apes using fluorescence in situ hybridization, Genomics 25:477–484, 1995. Return to text.
- Baldini, A. et al., An alphoid DNA sequence conserved in all human and great ape chromosomes: evidence for ancient centromeric sequences at human chromosomal regions 2q21 and 9q13, Human Genetics 90:577–583, 1993. Return to text.
- www.ncbi.nlm.nih.gov/. Return to text.


Readers’ comments
Comments are automatically closed 14 days after publication.