

Journal of Creation 20(2):82–89, August 2006
Browse our latest digital issue Subscribe
Do new functions arise by gene duplication?
Evolution requires a simple form of life to have morphed into increasingly complex organisms. Since the basis for biological complexity is genetic complexity, some biologists propose that the complicated genomes in modern organisms arose from one or a few genes in a common ancestor through duplication, with subsequent neofunctionalization through mutation and natural selection. Here we examine the known mechanisms of gene duplication in the light of genomic complexity and post-duplication events, and argue that: (1) gene duplications are aberrations of cell division processes and are more likely to cause malformation or diseases rather than selective advantage; (2) duplicated genes are usually silenced and subjected to degenerative mutations; (3) regulation of supposedly duplicated gene clusters and gene families is irreducibly complex, and demands simultaneous development of fully functional multiple genes and switching networks, contrary to Darwinian gradualism.

‘Natural selection merely modified, while redundancy created.’1
‘It might be said that all of the new genes arose from redundant copies of the pre-existed [sic] genes.’2
Regardless of how the first gene came into being, it is taught in textbooks that gene duplication is the major force driving evolution.3,4 Gene duplications do indeed add extra material to the genome, for example, by aberrations in the division of chromosomes during mitosis or meiosis, or by erroneous DNA replication. Evolutionists argue that with subsequent mutation and natural selection, one or all copies of a duplicated gene eventually encode new proteins (a process called ‘neofunctionalization’). Over millions of years, small simple genomes thus are believed to have evolved into large, complex ones, giving rise to the multiplicity of life forms both living and extinct.
One frequently cited evidence for gene duplication comes from gene sequence analyses. Sequence comparisons have revealed that some genes in modern organisms are more similar to each other than to other genes, and so they are classified into families. Gene families are especially abundant in large genomes. Family members within a genome, the paralogs, are believed to be products of gene duplications that have occurred in the past. Furthermore, functional domains of many proteins encoded by apparently unrelated genes also bear structural and functional similarities. All of these are used as evidence that the thousands of genes discovered so far (and those yet to be discovered) have evolved from a few—maybe one—ancestral gene(s).5
In this article we examine the major mechanisms proposed for gene duplication and evaluate their likely contribution to the history of life in the light of recent evidences on post-duplication events and gene regulation mechanisms.
Mechanisms of gene duplication
Polyploidy
Polyploidy refers to an increase in the number of sets of chromosomes per cell. Normally, most eukaryotic cells are diploid (with two sets of chromosomes, 2n, one from the male parent and one from the female parent) while the sex cells are haploid (with one set of chromosomes, 1n). A cell with 3n or more is polyploid. Polyploidy may arise naturally when a cell fails to divide after DNA replication. If the cell with doubled genome is involved in the generation of sex cells (meiosis), polyploid organisms may be subsequently produced upon fertilization. Alternatively, polyploidy can be artificially induced by treating cells with chemicals such as colchicine.
Since all genes are duplicated simultaneously in a polyploid cell, the stoichiometric relationships between genetic products are preserved. For this reason, polyploidy is the least detrimental and therefore the best surviving duplication mutation.6 Polyploidy is seen in ferns, flowering plants and some lower animals.7,8 It is usually associated with hermaphroditism, parthenogenesis (mother producing young asexually), or species without disparate sex chromosomes.8 In most dioecious (possessing either male or female organs) animals and humans, however, polyploid embryos typically suffer generalized malformation and die during development.8 It is not only sex determination per se (as was proposed by Muller9 ), but more importantly, the delicate balancing between homologous genes, that is disrupted in polyploid individuals of higher animals. For instance, parental imprinting (differences in the expression of maternal and paternal genes) by DNA methylation may be disrupted as the cell endeavours to silence extra chromosomes by extensive methylation (see below under ‘After duplication’).
Autopolyploidy (all chromosome sets are from the same species) can result in useful variation of quantitative traits such as biomass, organ size, flowering time, drought tolerance, etc. But crucially, polyploid organisms have an intrinsic mechanism to maintain genetic stability by silencing extra copies of genes (inhibiting their expression).10 Silencing of homeologs (genes duplicated by polyploidy) is nonrandom, genetically programmed, and organ-specific. It is a universal phenomenon seen in both plants and animals.7,11 Silencing of inferior alleles may be accountable for the advantageous phenotypes of some polyploid species. Alternatively, superior alleles may take dominance even though inferior ones are expressed simultaneously. In other words, there are no new genetic products, but old genes with altered expression levels under the control of pre-existing programs.

(b) Human globin gene clusters.53 Light grey: embryonic; Dark grey: fetal; Dark: fetal/adult (α) or adult only (δ and β); White: pseudogenes. Intergenic spacer sequences are omitted.
Allopolyploidy results when the sets of chromosomes are derived from two or more distinct, though related species. Unlike allodiploid hybrids such as the mule, allopolyploid organisms may be fertile and give rise to new species. However, the hybrid species display merely a new combination of pre-existing parental traits encoded by pre-existing genes. For example, some strains of the Triticale, synthetic allopolyploids from wheat and rye, combine the high yield of wheat and the adaptability of rye. Another artificial hybrid species between the tall fescue grass (Festuca arundinacea) and the short Italian ryegrass (Lolium multiflorum) shows quantitative traits (e.g. height) that are intermediate between the parental species.12 The historical Raphanobrassica, hybrid between cabbage and radish, has the roots of cabbages and leaves resembling that of a radish.
In allopolyploids there may be interactions between genes from different parents.13 Disharmonious interactions between homeologous genes are thought to be the reason for most cases of hybrid sterility in allodiploid animals.14 In plants, neoallopolyploid genomes are often unstable, displaying ‘sterility, lethality, and phenotypic instability’.15
Trisomy
In contrast to polyploidy, aneuploid cells (having a chromosome number that is not a multiple of the haploid) with one extra chromosome (trisomy) have a severely imbalanced genome. Consequently, the organism will manifest defective phenotypes. Aneuploidy is the result of failure to segregate a pair of homologous chromosomes during meiosis I or failure to segregate sister chromatids during meiosis II (meiotic nondisjunction). When a sex cell with one extra chromosome unites with a normal haploid sex cell, the zygote will be trisomic for that particular chromosome. Much knowledge about trisomy has been accumulated clinically in humans. Autosomal trisomies have more dramatic effects than sex chromosome trisomies. From the familiar Down syndrome (21 trisomy) to the less common Edward syndrome (18 trisomy) and Patau syndrome (13 trisomy), autosomal trisomies always hinder the development of the central nervous system and manifest mental retardation in live births. Developmental defects of other organs are also common. Trisomies involving other autosomes are rare, and are seen only in spontaneous abortions and in vitro fertilizations.16
Triplo-X females (karyotype XXX) have only mild symptoms (tallness and menstrual irregularities). While men with Klinefelter syndrome (karyotype XXY) show symptoms varying from infertility to severe structural deformation, XYY males are generally normal except for tallness and acne.17 The reason that sex chromosome trisomies show less severe symptoms than autosomal trisomies may lie in the fact that the X chromosome has a well established intrinsic inactivation mechanism to silence one homolog in the normal woman; while the Y chromosome is small with few genes.
Unequal crossing-over
Crossing-over refers to the exchange of fragments between homologous chromosomes during the initial stages of meiosis. Normally the exchange is equal as the genes line up based on sequence homology (synapsis). However, because of the numerous sequence repetitions in eukaryotic chromosomes, the lining up may be inaccurate, causing deletion in one chromosome and duplication in the other (figure 1). The mechanism is believed to be the major cause of deletions of red or green pigment genes in the X chromosome resulting in colour blindness and deletions of globin genes causing various forms of thalassemias.18,19 Repeated duplications have been associated with cancer.20 Duplication of a large segment of chromosome 15 in human beings can cause mental retardation and other symptoms while smaller duplications are asymptomatic or cause minor disorders such as panic attacks. Presumably, small segmental duplications are successfully managed by the cell’s silencing programs. However, segmental duplications within protein-coding sequences may interrupt gene structure, causing frame-shift mutations.21

Unequal crossing-over may have been the major mechanism in altering the number of genes in repetitive clusters. Gene clusters such as the human green pigment genes and the human immunoglobulin heavy chain genes that vary in numbers within the population certainly manifest recent duplications.22,23 Clusters of identical rRNA and histone genes also vary in number within the species, presumably via unequal crossing-over.24–28 Recently, it has been found that copy-number polymorphisms of this kind are more abundant than previously realized.29,30
However, it is unlikely that gene clusters originated through unequal crossing-over, because: (1) unequal crossing-over depends on pre-existing clustering. Although it may change the number of repetitions within clusters, unequal crossing-over is not the ultimate cause of their being; (2) multiplicity of identical genes in the clusters is often required for the cell to function properly. For instance, to meet the need of the cell to produce large numbers of ribosomes in a short time, all cells contain multiple copies of rRNA genes in tandem arrays. In the large oocyte (egg) of amphibians, the rRNA genes have to be further amplified approximately 2000-fold, resulting in about a million copies per cell, to maintain the number of ribosomes at about 1012.31 Likewise, multiple histone genes are required for the cell to synthesize histones rapidly during S phase of the cell cycle. But diversification and neofunctionalisation of these identical copies is actually prevented, not promoted, by as yet unknown mechanisms.32
Transposition
Transposons are mobile genetic elements that can change their positions within the genome (the process is known as transposition). While some transpositions occur by a ‘cut and paste’ mechanism, others go by a ‘copy and paste’ mechanism, resulting in duplications. Unlike unequal crossing-over that produces tandem gene arrays, transpositions cause duplications dispersed randomly throughout the genome. Transposons that duplicate via an RNA intermediate, known as retrotransposons, are abundant in eukaryotic cells.
Despite the abundance of transposons and retrotransposons in complex genomes (e.g. 45% of the human genome), their function remains elusive. Traditionally, they have been considered as ‘selfish DNA’ because random insertion of transposons disrupts other genes, causing deleterious mutations. A classical example is the Drosophila retrotransposon, the P element, which induces chromosomal breaks and causes sterility.33 Consequently, it seems to be beneficial to the organism for transposition events to be suppressed. Indeed, transposition is rare in the human cell. (Therefore, the vast majority of the human transposable elements must have been present in the genome since ancient times.) However, in mice, Drosophila (fruit-fly), and Arabidopsis (plant), transposition is still responsible for many mutations.34
Recently, Peaston and associates discovered that retrotransposons are actively transcribed in mouse oocytes and early embryos, providing alternative promoters and first exons to a subset of host genes.35 This report suggests that transposons function as regulatory elements during early development. From this point of view, transposition-induced mutation may be a side effect, instead of the intended function, of these repetitive genetic elements.
After duplication

In order for evolution to harness gene duplications to produce complex genomes, it was originally proposed that one or more copies of the duplicated gene will acquire advantageous mutations (neofunctionalization).5,36,37 This was thought to be the only mechanism to generate new genes from existing ones.38 However, biologists are now becoming more and more convinced theoretically and empirically that most duplicated gene copies undergo degenerative, rather than constructive, mutations, ending up in nonfunctionalization.
As stated above, the first event awaiting a duplicated gene is silencing. The best studied mechanism of silencing is through methylation of cytosine bases in CG islands around promoters.39 Subsequently, methylated cytosines tend to be spontaneously deaminated and are substituted with thymine bases.39,40 The phenomenon is known as CG depletion. Duplicated genes are especially prone to CG depletion.39–41 Without selective constraint, silenced duplicates may also undergo other mutations. Indeed, ‘extensive genomic change’ can be detected within a few generations after synthetic polyploidy.42 Using silent mutations (mutations that do not affect translated protein structures) to reflect time, Lynch and Conery calculated that duplicated genes are lost exponentially with time and are ‘nonfunctionalized by the time silent sites have diverged by only a few percent’.6
On the other hand, mutations in functioning gene family members are limited by purifying selection. In paralogous genes that evolutionists believe were created by ancient duplication events, ‘only about 5% of amino acid-changing mutations are able to rise to fixation’.6 There is a recent report that mutation rates in gene family members are actually lower than in singletons (genes without paralogs).43 In contrast, differences in amino acid sequences between modern paralogous genes are generally large, e.g. 58% between human α and β globins, 28% between human β and γ globins, 75% between human β globin and myoglobin.
Faced with this dilemma, some evolutionists theorized that mutations leading to neofunctionalization must have happened within a brief period of time immediately after duplication (in spite of the fact that the frequent mutations observed in recent duplicates are mostly degenerative).43 Realizing the impossibility of neofunctionalization, Lynch and Conery argued that gene duplication only passively contributes to the generation of biodiversity by building up reproductive barriers as duplicates are silenced stochastically.6 In other words, gene duplication does not produce new genes because silencing and subsequent degradation of duplicated genes cannot provide new information.
Meanwhile, several other models have been proposed concerning the fate of duplicated genes. One theory states that both the original and duplicated gene copies each lose only part of their function through degenerative mutations (subfunctionalization). If each gene copy retains a different fraction of its original function, the duplicates may complement each other and function together as one gene. If the regulatory elements of duplicated genes subfunctionalize (while the protein-coding regions are somehow spared from degeneration), they may be expressed at different stages/tissues. The theory is known as duplication-degeneration-complementation (DDC) model.44–46 The DDC model may allow partial preservation of duplicated genes, but it fails to explain the evolution of new genes or new regulatory elements. (Let alone the complicated mechanisms of tissue/organ-specific regulation. See below under ‘Gene Regulation’).
Recently, another model, called epigenetic complementation (EC), has been proposed by Rodin and colleagues.47,48 The theory states that if a gene is copied into a different position within the genome, it may be put under the control of a different regulatory environment and therefore expressed in a different tissue or stage of life. Epigenetic silencing mechanisms (such as cytosine methylation) work in such a way that one copy is silenced whenever or wherever the other copy is expressed. According to this model, there is no need for mutation to alter the regulatory elements of the duplicates in order to achieve complementation.
The EC model does not explain the existence of clustered gene families with diverged functions for each member. For example, the linked α and β globin genes in Xenopus laevis are expressed at different (tadpole and adult) stages of life (figure 2).49–51 But their temporal regulation is difficult to explain with differing epigenetic environments, since the adult genes are sandwiched between tadpole genes. Rather, it can be better accounted for by differences in their regulatory sequences that respond to stage-specific transcription factors.52,53 Similarly, members of the clustered human α globin gene family are expressed in two stages (embryonic and adult) and the clustered β globin gene family are expressed in three stages (embryonic, fetal, and adult) (figure 2). Again, temporal regulation (especially silencing) is accomplished genetically, rather than epigenetically, via distinct regulatory elements associated with the genes.54–56 Furthermore, there is no change in regulation of the globin genes after the supposed separation of α and β genes onto different chromosomes in mammals and birds. Both the ζ gene of the α family and the ε gene of the β family are expressed during the embryonic stage in human development, to form the ζ2 ε2 tetramer, even though they are on different chromosomes; while the α and β genes are expressed simultaneously in adults.
Like the DDC model, the EC model still depends on mutation and natural selection for neofunctionalization.
Genome complexity
If the evolution-by-gene-duplication theory is correct then DNA content and gene number should increase proportionately with organism complexity. However, this is not the case (Table 1). For example, the unicellular algae, Euglena, has a bigger genome than some vertebrate animals such as zebrafish and chicken. Amphibians may have genomes larger than some birds and mammals. The plant, Zea mays (corn), has more genomic DNA than does the human species. This phenomenon, known as the C-value paradox, demonstrates that the amount of genomic DNA is certainly not a good index for biological complexity.

Table 1 also shows that the number of genes within a genome does not increase in proportion to the amount of genomic DNA. As a general rule, larger genomes have sparser genes. Prokaryotic genomes are much more compact than eukaryotic genomes, e.g. 89% of Haemophilus genome consists of protein-coding genes as compared to 1–1.5% in the human genome. Consequently, the number of genes is an even poorer indicator of genome complexity than haploid DNA content. For example, human beings with 1014 cells have a total gene number comparable to that of Caenorhabditis elegans, which has only 959 somatic cells. Likewise, Drosophila, with its 50,000 cells, has only twice as many genes as the single-celled baker’s yeast.
In other words, simpler organisms already have DNA content and gene numbers comparable to that of advanced species. Further gene duplication (and mutation) will not help them climb up Darwin’s tree of life.
Gene regulation
Of course, it is not only the number of cells, but also the types of cells in an organism, that indicates complexity. On the genetic level, differentiation into various cell types is a result of the spatial and temporal regulation of genes. Therefore, the genes for transcription factors, which act as molecular switches in the genome, have much to do with genetic complexity. Prokaryotic genes are generally regulated as a group (polycistronic, i.e. several genes are controlled by one transcription factor) while eukaryotic genes are regulated individually (monocistronic).
Szathmary and associates proposed a mathematical formula to calculate genome complexity in terms of the interactions between genes (usually through their encoded protein products including transcription factors).60 He borrowed a parameter, connectivity (C), from ecology which uses the term to describe trophic interactions in food webs:
C = 2 L/[N(N-1)]
L refers to the number of interactions among genes (it originally meant ‘trophic links’ in ecology), while N refers to the number of genes in a genome (originally the number of species in an ecosystem). C is equal to the number of actual interactions out of all possible interactions.
The most important aspect of genetic interaction that determines the value of C in Szathmary’s equation is the number of levels constituting a regulation hierarchy. In ecosystems, adding trophic levels generates more connectivity than increasing the number of species. Like a food chain, a gene regulation pathway can have multiple levels of interactions, whereby upstream transcription factors regulate downstream transcription factors.
The concept of irreducible complexity61 applies to gene regulation systems. An irreducibly complex system is one in which all the essential parts must be present at the same time, and thus could not have been built up slowly over millions of years in a step-wise Darwinian fashion. In order for a gene regulation unit to function, many genetic elements, including trans-acting elements that encode the transcription factors, cis-acting elements that respond to the transcription factors, and the structural genes, have to be present simultaneously. Although there are examples of functional overlaps between pathways, multiple unique elements are usually required for each pathway. Knocking out any of the elements will frequently result in dysfunction, even loss of life.
In the simplest case, many viruses have three sets of genes regulated as a cascade (figure 3). The immediate-early (α) genes have promoter elements (binding sites for RNA polymerase or some transcription factors) similar to those of the host cell and are transcribed by a host cell RNA polymerase. The products of immediate-early genes are mostly transcription factors that interact with the cis-acting regulatory elements (promoter/enhancer) of early (β) genes. The early gene products, in their turn, activate the late (γ) genes, by interacting with their cis-acting elements. The early genes also encode enzymes to replicate the viral DNA, so that the late genes are multiplied before their expression, allowing for rapid accumulation of late gene products toward the end of infection. This scenario enables the virus to divert the resources of the host cell to the production of new viruses effectively.
A specific example of a regulation network is the major immediate-early gene (mIE) of the human cytomegalovirus (HCMV) which encodes two major products, IE1 and IE2, by alternative splicing (figure 4). The two proteins act synergistically to activate the β genes. Adjacent to the gene is a 1.1-Kb cis-regulatory sequence called the major immediate-early enhancer-promoter (MIEP), which contains concentrated binding sites for multiple cellular transcription factors. One of the products of the mIE gene, IE1, functions as an autoregulatory trans-activator that recruits a cellular protein, NF-kB, which binds to the enhancer and activates transcription. The IE2 product of the gene, on the other hand, represses the gene by binding to a cis-repression sequence (crs, see figure 4).62 The virus also carries several other viral proteins into the host cell for effective transcription of mIE. Among these are ppUL35 and pp71, which interact with each other in the infected cell.63,64 Meanwhile, pp71 interacts with a cellular protein, hDaxx, which is required for mIE transcription.65
Because the viral genome is relatively small and easy to manipulate, HCMV provides a good model in which to study the effects of knocking out a gene from the genome. Deletion of the sequences that encode IE2, or the proximal portion of the enhancer, from the HCMV genome completely inactivates the virus.66,67 Deletion of any of the genes that encode IE1, pp71, or ppUL35 renders the virus incapable of replication in vitro at low multiplicity of infection (MOI), which resembles natural human infection.68–70 All these regulatory factors have to be present and functional at the same time for HCMV to survive (if it cannot replicate it becomes extinct).
Virus genomes are far simpler in the complexity of their regulation than prokaryotes and eukaryotes, so it follows that their regulatory systems are also irreducibly complex. For evolution to have occurred via gene duplication, both the gene and its cis-regulatory elements have to be duplicated simultaneously. Furthermore, since gene family members often have distinctly different expression patterns, both the gene and the cis-regulatory elements have to mutate concertedly in order to confer a selective advantage to the organism. For example, the ζ and ε globins have to acquire higher oxygen affinity than the α and β globins in order for the embryonic hemoglobin tetramer ζ2 ε2 to extract oxygen from the maternal α2 β2 tetramers. Meanwhile, the regulatory elements of the embryonic and adult globins have to develop binding affinity for the transcription factors expressed during their respective developmental stages. Most importantly, a delicate globin switching mechanism, known to involve numerous trans-acting factors and multiple levels of regulation, has to be developed. In the case of the human β-like globin switching, which is the best understood, some of these factors are universal, while others are erythroid-specific.54–56,71 Deletion of the regulatory elements or a member of the gene family will result in thalassemia.
Another example of clustered gene families whose expression follows a temporal pattern is the immunoglobulin heavy chain family produced by B lymphocytes. There are five classes and each has properties that cannot be replaced by others. All B lymphocytes start by secreting IgM and switch to IgG, IgE, or IgA within a few days via a complex switching mechanism.72–74 The most important aspect of class switch is targeting of DNA recombination enzymes to specific sites. Gene duplication theory would require coordinated mutations in the structural genes and the cis-regulatory elements, and a unique recombination mechanism different from the known mechanisms.
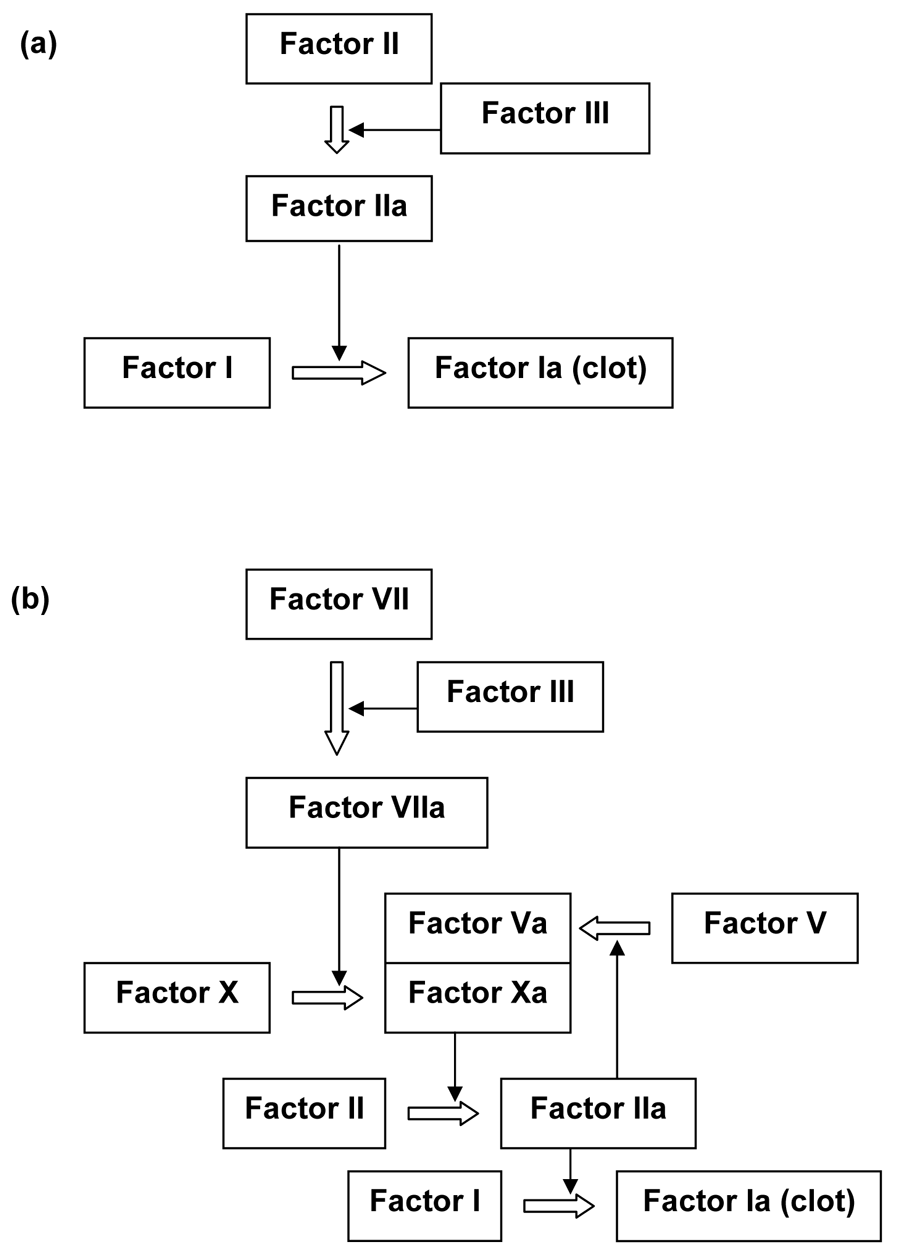
Michael Behe used the blood clotting factors to illustrate irreducible complexity.61 Dozens of proteins activate or inhibit each other in the blood coagulation and subsequent clot-dissolving pathways. Accidental deletion of factors leads to diseases such as hemophilia. Since many factors share similar functional domains, they are thought to have evolved by ancient gene duplication events, including polyploidy during the Cambrian explosion.75–77 However, these duplications have to be followed by coordinated mutations that ‘work just right’. A proposed functional intermediate blood clotting pathway75 in figure 5 shows how much coordinated change is required.
Conclusion
The majority of gene duplications are meiotic or mitotic aberrations, resulting in malformations or diseases. Plants can tolerate duplications, especially polyploidy, better than animals due to differences in their styles of reproduction. To maintain genomic stability, all cells have built-in mechanisms to silence duplicated genes, after which they become subject to degenerative mutations.
Clusters of identical genes need complicated mechanisms to prevent diversification in order for them to work in unison. Likewise, gene families whose members perform distinct functions are maintained by purifying selection. While duplication may alter the number of members in gene families, it is not their ultimate origin. Current models explaining the preservation and neofunctionalization of duplicated genes encounter obstacles one way or the other.
Evolution by gene duplication predicts a proportional increase in genome size with organism complexity but this is contradicted by the evidence. It is not genome size but intergenic regulatory sequences and gene regulation hierarchies that determine complexity. Gene regulation networks are irreducibly complex and constitute an insurmountable barrier for the theory.
References
- Ohno, S., Evolution by Gene Duplication, Spring-Verlag, Berlin, Heidelberg, New York, Preface, 1970. Return to text.
- Ohno, S., Birth of a unique enzyme from an alternative reading frame of the pre-existed, internally repetitious coding sequence, Proc. Natl. Acad. Sci.USA 81(8):2421–2425, 1984. Return to text.
- Hartwell, L.H., Hood, L., Goldberg, M.L., Reynolds, A.E., Silver, L.M. and Veres, R.C., Genetics, from Genes to Genomes, McGraw-Hill Companies, Inc., New York, pp. 449–452, 2004. Return to text.
- Lewin B., Genes VIII, Pearson Education, Inc., Upper Saddle River, NJ, p. 86, 2004. Return to text.
- Ohno, S., Evolution is condemned to rely upon variations of the same theme: the one ancestral sequence for genes and spacers, Perspect. Biol. Med. 25(4):559–572, 1982. Return to text.
- Lynch, M. and Conery, J.S., The evolutionary fate and consequences of duplicate genes, Science 290(5494):1151–1155, 2000. Return to text.
- Becak, M.L. and Kobashi, L.S., Evolution by polyploidy and gene regulation, Anura, Genet. Mol. Res. 3(2):195–212, 2004. Return to text.
- Otto, S.P. and Whitton, J., Polyploid incidence and evolution, Annu. Rev. Genet. 34:401–437, 2000. Return to text.
- Muller, H.J., Why polyploidy is rarer in animals than in plants, Am. Nat. 59:346–353, 1925. Return to text.
- Adams, K.L., Cronn, R., Percifield, R. and Wendel, J.F., Genes duplicated by polyploidy show unequal contributions to the transcriptome and organ-specific reciprocal silencing, Proc. Natl. Acad. Sci.USA 100(8):4649–4654, 2003. Return to text.
- Flavell, R.B., Inactivation of gene expression in plants as a consequence of specific sequence duplication, Proc. Natl. Acad. Sci.USA 91(9):3490–3496, 1994. Return to text.
- Brooker, R.J., Genetics, Analysis and Principles, McGraw-Hill Higher Eduction, New York, pp. 218–219, 2005. Return to text.
- Osborn, T.C., Pires, J.C., Birchler, J.A., Auger, D.L., Chen, Z.J., Lee, H.S., Comai, L., Madlung, A., Doerge, R.W., Colot, V. and Martienssen, R.A., Understanding mechanisms of novel gene expression in polyploids, Trends Genet. 19(3):141–147, 2003. Return to text.
- Stebbins, G.L. and Orr-Weaver,T.L., Polyploidy, in: AccessScience McGraw-Hill, <www.accessscience.com>, last modified: 10 April 2000. Return to text.
- Madlung, A., Tyagi, A.P., Watson, B., Jiang, H., Kagochi, T., Doerge, R.W., Martienssen, R. and Comai, L., Genomic changes in synthetic Arabidopsis polyploids, Plant J. 41(2):221–230, 2005. Return to text.
- Lewis R., Human Genetics, Concepts and Applications, McGraw-Hill Companies Inc., New York, pp. 246–249, 2003. Return to text.
- Lewis, ref. 16, pp. 249–251. Return to text.
- Hartwell et al., ref. 3, pp. 226–227. Return to text.
- Lewin, ref. 4, pp. 96–97. Return to text.
- Lucito, R., Healy, J., Alexander, J., Reiner, A., Esposito, D., Chi, M., Rodgers, L., Brady, A., Sebat, J., Trope, J., West, J.A., Rostan, S., Nguyen, K.C., Powers, S., Ye, K.Q., Olshen, A., Venkatraman, E., Norton, L. and Wigler, M., Representational oligonucleotide microarray analysis: a high-resolution method to detect genome copy number variation, Genome Res. 13(10):2291–2305, 2003. Return to text.
- O’Dushlaine, C.T., Edwards, R.J., Park, S.D. and Shields, D.C., Tandem repeat copy-number variation in protein-coding regions of human genes, Genome Biol. 6(8):R69, 2005. Return to text.
- Nathans, J., Thomas, D. and Hogness, D.S., Molecular genetics of human color vision: the genes encoding blue, green and red pigments, Science 232(4747):193–202, 1986. Return to text.
- Rabbani, H., Pan, Q., Kondo, N., Smith, C.I. and Hammarstrom, L., Duplications and deletions of the human IGHC locus: evolutionary implications, Immunogenetics 45(2):136–141, 1996. Return to text.
- Zhang, Q.F., Saghai Maroof, M.A. and Allard, R.W., Effects on adaptedness of variations in ribosomal DNA copy number in populations of wild barley (Hordeum vulgare ssp. spontaneum), Proc. Natl. Acad. Sci.USA 87(22):8741–8745, 1990. Return to text.
- Bentley, R.W. and Leigh J.A., Determination of 16S ribosomal RNA gene copy number in Streptococcus uberis, S. agalactiae, S. dysgalactiae and S. parauberis, FEMS Immunol. Med. Microbiol. 12(1):1–7, 1995. Return to text.
- Su, M.H. and Delany, M.E., Ribosomal RNA gene copy number and nucleolar-size polymorphisms within and among chicken lines selected for enhanced growth, Poult. Sci. 77(12):1748–1754, 1998. Return to text.
- Michel, A.H., Kornmann, B., Dubrana, K. and Shore, D., Spontaneous rDNA copy number variation modulates Sir2 levels and epigenetic gene silencing, Genes. Dev. 19(10):1199–1210, 2005. Return to text.
- Thomas, M.C., Olivares, M., Escalante, M., Maranon, C. and Montilla, M., Plasticity of the histone H2A genes in a Brazilian and six Colombian strains of Trypanosoma cruzi, Acta. Trop. 75(2):203–210, 2000. Return to text.
- Sebat, J., Lakshmi, B., Troge, J., Alexander, J., Young, J., Lundin, P., Maner, S., Massa, H., Walker, M., Chi, M., Navin, N., Lucito, R., Healy, J., Hicks, J., Ye, K., Reiner, A., Gilliam, T.C., Trask, B., Patterson, N., Zetterberg, A. and Wigler, M., Large-scale copy number polymorphism in the human genome, Science 305(5683):525–528, 2004. Return to text.
- Sharp, A.J., Locke, D.P., McGrath, S.D., Cheng, Z., Bailey, J.A., Vallente, R.U., Pertz, L.M., Clark, R.A., Schwartz, S., Segraves, R., Oseroff, V.V., Albertson, D.G., Pinkel, D. and Eichler, E.E., Segmental duplications and copy-number variation in the human genome, Am. J. Hum. Genet. 77(1):78–88, 2005. Return to text.
- Cooper, G.M. and Hausman, R.E., The Cell, a Molecular Approach, ASM Press, Washington, DC, pp. 339–341, 2004. Return to text.
- Lewin, ref. 4, pp. 100–103. Return to text.
- Lewin, ref. 4, pp. 487–490. Return to text.
- Cooper and Hausman, ref. 31, p. 225. Return to text.
- Peaston, A.E., Evsikon, A.V., Graber, J.H., de Vries, W.N., Holbrook, A.E., Solter, D. and Knowles, B.B., Retrotransposons regulate host genes in mouse oocytes and preimplantation embryos, Dev. Cell 7(4):597–606, 2004. Return to text.
- Ohno, ref. 1, pp. 71–87. Return to text.
- Taylor, J.S. and Raes, J., Duplication and divergence: the evolution of new genes and old ideas, Annu. Rev. Genet. 38:615–643, 2004. Return to text.
- Ohno, ref. 1, p. 2. Return to text.
- Lewin, ref. 4, pp. 620–621. Return to text.
- Lund, G., Lauria, M., Guldberg, P. and Zaina, S., Duplication-dependent CG suppression of the seed storage protein genes of maize, Genetics 165(2):835–848, 2003. Return to text.
- Rodin, S.N. and Parkhomchuk, D.V., Position-associated GC asymmetry of gene duplicates, J. Mol. Evol. 59(3):372–384, 2004. Return to text.
- Song, K., Lu, P., Tang, K. and Osborn, T.C., Rapid genome change in synthetic polyploids of Brassica and its implications for polyploid evolution, Proc. Natl. Acad. Sci.USA 92(17):7719–7723, 1995. Return to text.
- Jordan, I.K., Wolf, Y.I. and Koonin, E.V., Duplicated genes evolve slower than singletons despite the initial rate increase, BMC Evol. Biol. 4:22, 2004. Return to text.
- Force, A., Lynch, M., Pickett, F.B., Amores, A., Yan, Y.L. and Postlewait, J., Preservation of duplicate genes by complementary, degenerative mutations, Genetics 151(4):1531–1645, 1999. Return to text.
- Lynch, M. and Force, A., The probability of duplicate gene preservation by subfunctionalization, Genetics 154:459–473, 2000. Return to text.
- Lynch, M., O’Hely, M., Walsh, B. and Force, A., The probability of preservation of a newly arisen gene duplicate, Genetics 159:1789–1804, 2001. Return to text.
- Rodin, S.N., Parkhomchuk, D.V. and Riggs, A.D., Epigenetic changes and repositioning determine the evolutionary fate of duplicated genes, Biochemistry (Mosc.) 70(5):559–567, 2005. Return to text.
- Rodin, S.N. and Riggs, A.D., Epigenetic silencing may aid evolution by gene duplication, J. Mol. Evol. 56(6):718–729, 2003. Return to text.
- Banville, D. and Williams, J.G., The pattern of expression of the Xenopus laevis tadpole alpha-globin genes and the amino acid sequence of the three major tadpole alpha-globin polypeptides, Nucleic Acids Res. 13(15):5407–8421, 1985. Return to text.
- Hosbach, H.A., Wyler, T. and Weber, R., The Xenopus laevis globin gene family: chromosomal arrangement and gene structure, Cell 32(1):45–53, 1983. Return to text.
- Patient, R.K., Banville, D., Brewer A.C., Elkington, J.A., Greaves, D.R., Lloyd, M.M. and Williams, J.G., The organization of the tadpole and adult alpha globin genes of Xenopus laevis, Nucleic Acids Res. 10(24):7935–7945, 1982. Return to text.
- Broyles, R.H., Ramseyer, L.T., Do, T.H., McBride, K.A. and Barker, J.C., Hemoglobin switching in Rana/Xenopus erythroid heterokaryons: factors mediating the metamorphic hemoglobin switch are conserved, Dev. Genet. 15(4):347–355, 1994. Return to text.
- Cooper and Hausman, ref. 31, p. 148. Return to text.
- Sargent, T.G., Buller, A.M., Teachey, D.T., McCanna, K.S. and Lloyd, J.A., The gamma-globin promoter has a major role in competitive inhibition of beta-globin gene expression in early erythroid development, DNA Cell Biol. 18(4):293–303, 1999. Return to text.
- Harju, S., McQueen, K.J. and Peterson, K.R., Chromatin structure and control of beta-like globin gene switching, Exp. Biol. Med. (Maywood) 227(9):683–700, 2002. Return to text.
- Ristaldi, M.S., Drabek, D., Gribnau, J., Poddie, D., Yannoutsous, N., Cao, A., Grosveld, F. and Imam, A.M., The role of the–50 region of the human gamma-globin gene in switching, EMBO. J. 20(18):5242–5249, 2001. Return to text.
- Cooper and Hausman, Ref. 31, p. 12. Return to text.
- Lynch, M. and Conery, J.S., The origins of genome complexity, Science 302(5649):1401–1404, 2003. Return to text.
- International human genome sequencing consortium, Finishing the euchromatic sequence of the human genome, Nature 431:931–945, 2004. Return to text.
- Szathmary, E., Jordan, F. and Pal, C., Molecular biology and evolution. Can genes explain biological complexity? Science 292(5520):1315–1316, 2001. Return to text.
- Behe, M.J., Darwin’s Black Box, The Free Press, New York, 1996. Return to text.
- Mocarski, E.S.J., Cytomegaloviruses and their replication; in: Field, B.N., Knipe, D.M., Howley, P.M., Chanock, R.M., Melnick, J.L., Monath, T.P., Roizman, B. and Straus, S.E. (Eds.), Fields Virology, 3rd ed., Lippincott-Raven Publishers, Philadelphia, PA, pp. 2447–2492, 1996. Return to text.
- Liu, Y. and Biegalke, B.J., The human cytomegalovirus UL35 gene encodes two proteins with different functions, J. Virol. 76:2460–2468, 2002. Return to text.
- Schierling, K., Stamminger, T., Mertens, T. and Winkler, M., Human cytomegalovirus tegument proteins ppUL82 (pp71) and ppUL35 interact and cooperatively activate the major immediate-early enhancer, J. Virol. 78:9512–9523, 2004. Return to text.
- Hoffmann, H., Sindre, H. and Stamminger, T., Functional interaction between the pp71 protein of human cytomegalovirus and the PML-interacting protein human Daxx, J. Virol. 76(11):5769–5783, 2002. Return to text.
- Marchini, A., Liu, H. and Zhu, H., Human cytomegalovirus with IE-2 (UL122) deleted fails to express early lytic genes, J. Virol. 75(4):1870–1878, 2001. Return to text.
- Isomura, H., Tsurumi, T. and Stinski, M.F., Role of the proximal enhancer of the major immediate-early promoter in human cytomegalovirus replication, J. Virol. 78(23):12788–12799, 2004. Return to text.
- Ghazal, P., Visser, A.E., Gustems, M., Garcia, R., Borst, E.M., Sullivan, K., Messerle, M. and Angulo, A., Elimination of ie1 significantly attenuates murine cytomegalovirus virulence but does not alter replicative capacity in cell culture, J. Virol. 79(11):7182–7194, 2005 Return to text.
- Bresnahan, W.A. and Shenk, T.E., UL82 virion protein activates expression of immediate early viral genes in human cytomegalovirus-infected cells, Proc. Natl. Acad. Sci.USA 97:14506–14511, 2000. Return to text.
- Schierling, K., Buser, C., Mertens, T. and Winkler, M., Human cytomegalovirus tegument protein ppUL35 is important for viral replication and particle formation, J. Virol. 79(5):3084–3096, 2005. Return to text.
- Zhao, Q., Zhou, W.L., Rank, G., Sutton, R., Wang, X., Cumming, H., Cerruti, L., Cunningham, J.M. and Jane, S.M., Repression of human {gamma}-globin gene expression by a short isoform of the NF-E4 protein is associated with loss of NF-E2 and RNA polymerase II recruitment to the promoter, Blood, 1 November 2005; [Epub ahead of print]. Return to text.
- Okazaki, I.M., Kinoshita, K., Muramatsu, M., Yoshikawa, K. and Honjo, T., The AID enzyme induces class switch recombination in fibroblasts, Nature 416(6878):340–345, 2002. Return to text.
- Luby, T.M., Schrader, C.E., Stavnezer, J. and Selsing, E., The mu switch region tandem repeats are important, but not required, for antibody class switch recombination, J. Exp. Med. 193(2):159–168, 2001. Return to text.
- Mills, F.C., Harindranath, N., Mitchell, M. and Max, E.E., Enhancer complexes located downstream of both human immunoglobulin Calpha genes, J. Exp. Med. 186(6):845–858, 1997. Return to text.
- Davidson, C.J., Tuddenham, E.G. and McVey, J.H., 450 million years of hemostasis, J. Thromb. Haemost. 1(7):1487–1494, 2003. Return to text.
- Jiang Y. and Doolittle, R.F., The evolution of vertebrate blood coagulation as viewed from a comparison of puffer fish and sea squirt genomes, Proc. Natl. Acad. Sci.USA 100(13):7527–7532, 2003. Return to text.
- Naruse, K., Tanaka, M., Mita, K., Shima, A., Postlethwait, J. and Mitani, H., A medaka gene map: the trace of ancestral vertebrate proto-chromosomes revealed by comparative gene mapping, GenomeRes. 14(5):820–828, 2004. Return to text.
Readers’ comments
Comments are automatically closed 14 days after publication.