

Journal of Creation 26(1):76–85, April 2012
Browse our latest digital issue Subscribe
The proportion of polypeptide chains which generate native folds—part 5: experimental extraction from random sequences
This is the second to last of a six part series which critiques the most widely cited papers claiming that a high proportion of random polypeptides naturally form native-like folds. Parts 5 and 6 might not seem like exciting reading, but they cover the only known experiments we are aware of which look at random polypeptides. When Professor Robert Sauer from MIT was contacted by the author questioning some published claims, no effort was made to defend his own work. Instead, attention was drawn to the research by Professor Jack Szostak, which is discussed here.
We find the claim that one out of 1011 random polypeptides in free nature would produce proteins reliably folded to be unconvincing. The artificial polypeptides identified were much smaller than average-sized proteins, depend on the presence of zinc and ATP, and lacked the rich secondary structures characteristic of biological proteins.
What proportion of random polypeptide chains based on the twenty natural amino acids would fold into native-like folds? Ideally, one would examine experimentally a library of random sequences to answer this question. However, examining a vast number of random sequences 150 AAs (amino acids) long (the average size of a domain)1 or 300 AA (the average size of a protein) is not feasible should indeed the proportion of those folding reliably be very small. Experimental methods such as CD (circular dichroism) spectra, NMR, denaturating spectra, X-ray crystallography, and so on, are very time-consuming.
To our knowledge only the series of experiments pioneered by Professor Szostak, winner of the 2009 Nobel Prize in Physiology or Medicine, have addressed this question empirically. During the last decade other researchers had made contributions to this original work, and some have concluded that a fairly high proportion of random polypeptides can fold stably. Lo Surdo states that
“It had been previously suspected that the probability of a stable fold arising at random is extremely small (argument based on the complexity paradox that the disparity between the many potential protein sequences and the relatively few different structures known). However, we have confirmed that functionally directed in vitro evolution from random sequences can generate a novel fold with a tailored function. In addition, such folds may display the key features of naturally evolving proteins.”2
We shall examine in this paper two key studies and two more in the next part of this series to see if this conclusion is warranted.
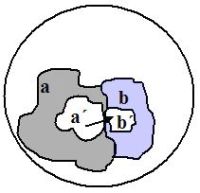
The proportion of native-like folded polypeptides is expected to decrease as chain size increases, due to undesirable interferences and decreasing solubility. For this reason, these experiments typically work with small chains of n = 80 AA (or shorter). The potential sequence space therefore covers 20n = 2080, or 1 × 10104 alternative chains. Through clever search strategies, shown in figure 1, the researchers iteratively narrow the search space to promising regions in the sequence space.
First study
A method to create large libraries of mRNA sequences lacking frameshifts, stop codons and internal translation initiating events has been developed3 and was combined with the experimental technology described under ‘Methods’ below. Typically about 6 × 1012 different sequences are rapidly examined in these experiments based on their ability to bind to immobilized ATP in a separation column. The researchers reasoned that strong and selective binding to ATP might be a relevant criteria to identify properly folded proteins. Noteworthy is that ATP binding proteins are found in all major enzyme classes.4

Since 1012 << 2080, the maximum number of alternative sequences 80 AA long, no duplicate sequences were likely to have been generated. After removing the sequences which did not bind to immobilized ATP, a subset of promising candidates were isolated (members a´ in figure 1). Error-prone PCR amplification generated new, similar variants (region b in figure 1) likely not to have been present among those in sampling space a. From these the best candidates were isolated (sequences b´, figure 1). This process was repeated to select other polypeptides with yet stronger affinity for ATP, sequences c´, and so on. In this manner, promising portions of the overall search space can be efficiently identified.
The key insight is that sequences b´ were the result of an intelligently directed search, and have a high probability of including sequences able to bind better to ATP than the best from a´ could. They are members of a much larger pool of candidates, a + b (figure 1).
The search strategy assumes that sequences very different from those producing native-like folded proteins could be initially identified by natural selection and thereafter improved upon. This view is illustrated as assumption ‘A’ in figure 2. However, others5 believe that chains significantly different from native folds are useless as an evolutionary starting point, since there are countless energy minima which have no relevance to the protein folding state (assumption ‘B’ in figure 2). This matter is not addressed in these series of studies, although clearly the authors simply assume ‘A’ to be true.
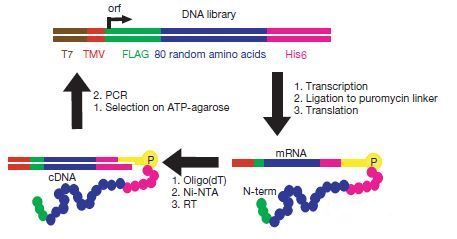
Now to the specifics. In the seminal paper, Keefe and Szostak6 prepared a library of 6 × 1012 non-redundant random proteins 80 AA long, designed to avoid random stop codons so as to generate the maximum variety possible.7 The random 80 AA region was flanked by short affinity tags to permit subsequent purification of the proteins, figure 3.
The eluted fractions were amplified by polymerase chain reaction (PCR). The DNA produced was used to generate another library of mRNA-displayed proteins and the scheme repeated for a total of eight rounds (figure 3). During the first round about 0.1% of the proteins bound to ATP in the column, and after eight rounds of selection the amount had increased to 6.2%. The low level of ATP-binding of the best sequences isolated so far (6.2%) is probably due to the conformational heterogeneity of the sequences isolated7: the same sequence can fold in many non-native-like manners.
The authors cloned and sequenced 24 of the members after the eighth round and found that most could be grouped into four sequences (figure 4A). These were unrelated to each other and to any biological sequences known. Representatives of each of the four families (A–D) were examined. Between 5% and 15% of these fused mRNA-displayed proteins bind to immobilized ATP (see last column of figure 4A). Note that far more of the chain binds to ATP if still attached to its mRNA (second to last column of figure 4A), which was not discussed in the publication. This fact illustrates that organic molecules such as ATP can easily bind to many classes of bio-chemicals, not just to polypeptides.
In order to optimize binding to ATP, the improved library was mutagenized three times using mutagenic PCR amplification, with an average rate of 3.7% per amino acid for each round. After six additional rounds of amplification without mutagenesis and in vitro selection followed by elution with ATP, the proportion which binds to ATP in the column rose to 34%.
Of the 56 clones sequenced, all were derived from a member of the family B (figure 4B).

|

|

|

|
Figure 4. A) Consensus sequences of initially selected protein families after round 8; B) Sequence data of mutagenized re-selected proteins after round 18, free protein; C) Truncated versions of DNA-tagged protein ‘18–19’; D) Deletion analysis of clone 18–19 fused to MBP protein.12 Click here for a larger image. |
Comparing to the original family B revealed that the mutations generated had produced four which bound effectively to ATP (present more than 39 times in the 56 sequences examined), see second row from the top in figure 4b, labelled ‘18predom’. In addition, 16 other substitutions generated were also selectively enriched (present more than 4 times among the 56 examined), see second row from the top in figure 4b, labelled ‘18select’. This suggests that different amino acids distributed throughout the protein sequence might be interacting with various portions of ATP.
After selection in round 18, eight individual proteins were chosen randomly. As free protein, the proportion which binds in the column (and was then eluted with free ATP) varied from 5% to 40%.12
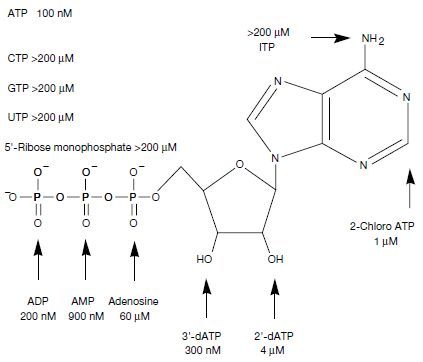
The four proteins that bound best to ATP were expressed in E. coli as fused proteins to maltose-binding protein (MBP), which is often done when the proteins are not soluble enough to work with conveniently. The clone which bound best was labelled ‘18–19’ (figure 5) and had a dissociation constant of Kd = 100 nM for ATP at 4°C and 1:1 stoichiometry. However, gel filtration indicated that 65% of 18–19 is monomeric and the rest binds in higher-order aggregates. Only the monomers bond to ATP.
Unsurprisingly, selection for binding to ATP did not improve binding to various other bio-chemicals. Chemical analogs to ATP, such as CTP13, GTP14, UTP15, and ITP16 bound far less effectively to protein 18–19 (figure 5). Furthermore, removing one, two or three phosphate groups led to molecules which bound less effectively to ATP (figure 5). Removing either of the 2´ or 3´ hydroxyl groups also reduced the amount of binding.
The minimal region for ATP binding of clone 18–19 was explored by deleting portions of the protein. The fraction of the protein which bound to the ATP in the column, relative to 18–19, is shown in the last column of figure 4C. Removing large sections from 18–19 improved the binding: those residues were apparently interfering with binding to ATP. A core domain of 45 amino acids was found to be sufficient to bind efficiently.12 Significantly, the deletion shown in the fourth row from the top in figure 4C had a ten-fold deleterious effect on ATP binding. This drew attention to the fact that a CXXC motif, involving two cysteine amino acids, had been destroyed.
In another experiment, variant 18–19 was fused to MBP protein and the dissociation constants, Kd, were measured after removing various fragments (figure 4D). Loss of affinity was observed for sections removed in the N-terminal portion (left-hand side of the sequences in figure 4D, note the last three rows). The authors propose that regions surrounding the important core which interacts with ATP might stabilize its structure or that additional amino acids might also interact with ATP.12 This observation is consistent with the accepted view that domains require a significant number of residues to be present. The CATH database of domains gives an average domain length of 159.5 AAs and the three smallest have 13, 14, and 17 AAs (out of 128,688 entries). These three smallest domains consist of merely a single tiny alpha coil.18At the large end, the CATH database revealed 18 domains having over 500 AAs.18

The authors concluded, on the basis of their experiments that
“The frequency with which ATP-binding proteins occur in sequence space can be estimated from the observed recovery of four such proteins from a non-redundant library of 6 x 1012 random sequences. On the basis of the average behaviour of the proteins isolated before mutagenesis, only about 10% of the potentially functional sequences present in the first round would be expected to generate correctly folded active proteins and thus survive to be amplified … We therefore estimate that roughly 1 in 1011 of all random sequence proteins have ATP-binding activity comparable to the proteins isolated in this study.”
Four years after this work3 was published, the crystal structure of protein 18–19 was elucidated2,19 (figures 6A–C) independently by two groups, and it was reported20 to overlap quite well with a small portion of domain ARG-GAP of a biological protein (figure 7).
It must be emphasized that ADP, and not ATP, was found co-crystallized with clone 18–19, even when the presence of ADP was rigorously prevented.
Second study
In a similar study23 also performed in Dr Szostak’s lab (see ‘Methods’ below), polypeptides able to bind to streptavidin24 (figure 8) were identified from a semi-random library of 6.7 × 1012 members.

The library had been designed using short eleven-amino-acid cassettes which were then concatamerized together. By design, 44% of the incorporated cassettes encoded a peptide polar/non-polar pattern compatible to forming amphipathic alpha-helices and 45% to form beta-strands; 11% were not patterned. Similar experiments to that described above were performed6 except that mutations were not deliberately generated. After seven rounds of selection for streptavidin and amplification with PCR, 20 different sequences were observed and analyzed. All were found to have frame-shifted, destroying the intended patterns which would lead to secondary structure. This experimental misfortune does provide us with another example of random sequences, however. The mutations and subsequence strong selection were due to the generation of one or more of the amino acid pattern HPQ,27 which is known to bind strongly to streptavidin.
The clone with a single HPQ pattern which bound most strongly to streptavidin was selected for additional study. Removing more than half of the residues led to a 38-amino-acid species with slightly higher binding affinity, but additional deletions lowered the affinity.
The authors provide two possible explanations for these observations:
- The flanking amino acids might stabilize the active conformation of the HPQ tripeptide motif.
- Several distinct peptide elements could be interacting with different parts of the streptavidin.
Evaluation of the first study
The B-family proteins possess a pair of CXXC motifs (two Cysteines separated by two amino acids, ‘X’) (highlighted in figures 4A–D) and the presence of a single bound zinc, revealed by atomic absorption spectroscopy. The proteins with good ATP-binding properties, including the best-binding clone 18–19, require zinc. The authors suggested that these proteins
“… bind to ATP with a folded structure nucleated around, or stabilized by, a Zn2+ ion coordinated to the four invariant cysteines of the CXXC sequences.”17
They reported that Mg2+ and other cations were not found to be suitable. Note that
“Zinc was not added to the selection buffer, but is present at about 10 micromole mammalian blood, from which the reticulocyte lysate used for mRNA translation is prepared.”17
The presence of zinc was accidental and no proteins were reported which bound well to ATP which lacked this metal.
As reiterated in a later paper,28 the estimate of 1 out of 1011 involved sequences for which
“All of the variants examined required high concentrations of free ATP in order to remain stably folded and soluble.”29
They add,
“… the ancestral and binding optimised proteins formed visible precipitates during the first 24 h and became completely insoluble after three days. Surprisingly, the core domain of the divergent protein 18–19 also formed a visible precipitate after 24 h and was almost completely insoluble by three days.”30
In the process of preparing 18–19 for X-ray diffraction, large amounts were expressed in E. coli and then purified to homogeneity. Apparently the natural mixture of 18–19 conformations was not used:
“Our structure determination targeted the functional core of ANBP [ANBP is an abbreviation for ‘artificial nucleotide binding protein’]. We aimed to optimise the production of a protein with a single conformation by screening different ANBP constructs, thus identifying N- or C-terminal residues unnecessary for either folding or ligand binding.”31
The implication is that the secondary structure shown in the crystals generated seem not to represent the conformation formed in the solvated state at biologically relevant temperatures.
Only residues 7–73 could be identified in the electron density map, and residues 7–10 on the N-terminus were poorly defined.32 The X-ray data revealed considerably smaller and less secondary structure than observed in CATH biological domains classified as Alpha Beta.33 The crystals were developed at 100 K, a very low temperature, far below the freezing point of water. These facts suggest that under ambient conditions very little, if any, secondary structure is actually present. It is well known that the crystal state of a protein sometimes does not reflect its topology in solution.34 Incidentally, the N-terminal alpha helix had been provided by amino acids present in the FLAG sequence, and therefore cannot be legitimately considered representative of a random sequence. Removing this alpha helix from figure 6B leaves still less secondary structure, considerably less than found in virtually all native proteins.
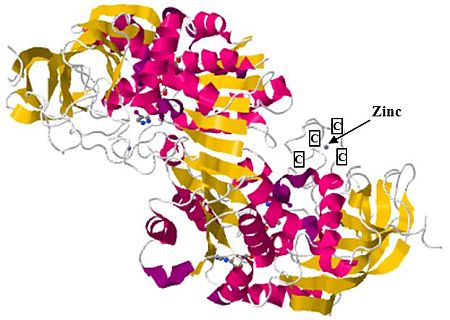
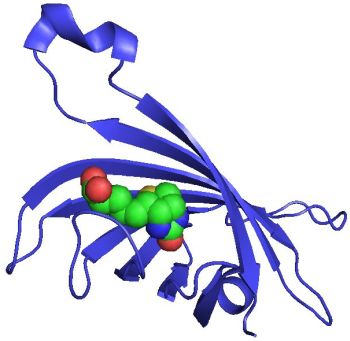
Figure 6A shows how the four cysteines binding to zinc hold the secondary structures together. However, this feature is not observed in biological proteins for which a similar zinc motif has been found (figure 7): the region held together by the zinc atom, while surely of biological value, is not responsible for the folding of the large domain. Indeed, the tiny zinc-chelated portion is far removed from the alpha coils and beta sheets, best seen by rotating the molecule using a protein viewer such as RasTop.
A well-known protein motif is known as the zinc finger.35 These are very small structures found on about 500 proteins in humans,36 which usually consist of two cysteines located in a short strand or turn region followed by an α-helix which contains two His ligands. The key observation is that although zinc is used with these biological motifs, the zinc fingers are not responsible for the folded structure of the proteins in which they are found.
Figure 9 illustrates another example where zinc is chelated to four cysteins in a representative biological protein with rich secondary structure. Notice how the alpha coils and beta sheets are very distant from the chelation site, so that the native fold could not be driven by the presence of zinc.
However, in the synthetic protein described, a zinc atom seems to be entirely responsible for what little structure is present in the protein.
Analysis of the second study
We offer the following comments about this second study:
- For the optimized 38-amino acid polypeptide, 13% did not bind even at high concentrations of streptavidin. Real proteins involved in enzymatic catalysis are highly specific and could not fail to bind to their intended ligands under these experimental conditions.
- Removing more than half of the polypeptide led to slightly stronger binding, which reflects the potential for larger chains to offer steric interference to protein– ligand interactions. This reinforces the fact that as chain size for random polypeptides increases, the probability of deleterious interferences and insolubility increase.
- Replacing the HPQ pattern with a similar HGA reduced the extent of binding by a factor of a thousand.37 The special HPQ pattern was almost the whole cause for binding to streptavidin.38 This reinforces the need to be very close to the final and correct sequence before natural selection can sense the gene’s existence.
Evaluation of these studies
As reported in a later contribution, “Unfortunately, biophysical characterization of the selected ATP binding proteins proved impossible due to poor solubility.”39 For these reasons, laboratory tests necessary to characterize the secondary and tertiary structure, such as CD spectra and NMR, were not available when the first study was published.6
Analysis of the data published until 2004 suggests the following interpretation. The fortuitous contamination with zinc has led to a chelating site, thanks to the presence of CXXC motifs. Some of the isomers were thereby able to offer a small surface which can be moulded in the presence of ATP to permit some as of yet uncharacterized interactions. Notice how ATP only seems to interact with a single tiny beta sheet, see figure 6B. Selection for ATP did not generate a microenvironment on the protein which was also conducive to strong binding of ATP chemical analogs such as CTP, GTP, UTP, and ITP. This is not particularly surprising, given the selective protocol.
Removal of portions of the protein and improved binding upon mutating various positions need not indicate multiple interactions, the strength of which has been improved, but rather the removal of disturbing amino acids which can lead to insolubility, entanglement, intermolecular interactions and such features deleterious to ATP binding.
Often the structure of a protein in its dissolved condition under natural conditions and temperature is very different from the structure elucidated after crystallization using X-ray diffraction. The process of crystallizing clone 18–19, at very cold temperatures, can select the particular conformations which are particularly stable. We suspect that the minimal secondary structure identified (figure 6B) is an artefact of the crystallization. Chelation with zinc forces several amino acids into close proximity, facilitating creation of alpha coils and beta sheets under biologically irrelevant conditions.
The limited data available supports this view. In the next part to this series, we draw attention to later work in which the authors discovered, unexpectedly, that CD spectroscopy at biologically relevant temperatures denied the presence of secondary structure. In addition, we must draw attention to the fact that in the absence of ATP suitable crystals were not formed. It appears that not only zinc, but also ATP is responsible for moulding clone 18–19, since otherwise it would not crystallize. The evidence is clear that this protein alone cannot produce a native-like fold under the above water-freezing temperatures. In a later paper we read,
“Unlike many naturally occurring proteins, protein 18–19 requires high concentrations of free ligand in order to remain stably folded and soluble.”40
This fact was not mentioned in the earlier work, nor quotes made subsequently of the estimated proportion of random peptides with native-like folds.
We welcome these kinds of studies to test the plausibility of our models, whether one believes in creation or evolution. We have some suggestions for those involved in this kind of work.
- Justify the belief that highly specific binding to a ligand such as ATP, but not analogs, can permit an estimate of random proteins which produce a native-like fold. Why must a native-like fold be able to bind to this particular molecule? Might not additional ligands also bind strongly to other members of the sampled portion? Why must a strong interaction at a judiciously constrained portion of the protein have any relevance to native protein folding?
- Generate a large number of variants similar to clone 18–19 and then determine whether there is evidence for secondary and native structure in the absence of ATP and zinc.
- Repeat the studies based on selection for ATP or another ligand, using n = 40 … 200 amino acids, to determine whether the proportion (not absolute number) with a given level of binding follows a pattern. If larger random-sequence proteins become more chaotic, multimeric and insoluble, and prevent binding to a ligand, then native-like folds will be less likely.
We offer some objections for the suggested proportion of random polypeptides leading to stable folding, 10–11, for evolutionary modelling purposes.
- The data suggests that ATP is necessary, but once bound to the crystallized clone 18–19 was immediately destroyed (hydrolyzed to ADP). Since ATP has negative charge, it is clear that it binds to the basic amino acids, as seen in 18–19 and other crystal structures. ATP has many hydrogen and oxygen atoms which are able to form numerous H-bonds. And adenine in ATP can form planar stacking interactions with aromatic residues. No wonder that it binds somewhere! Zinc can bind to cysteines and other residues41,42 such as the side chains of aspartic acid, glutamic acid and histidine, leading to local structures able to bind bio-molecules. These facts illustrate the vast number of destructive possibilities available along an evolutionary path towards forming a properly folded protein. And using an example which indiscriminately destroys the eminently valuable energy carrier, ATP, is a questionable argument for how proteins could arise naturalistically. It appears rather to highlight constraints which must be overcome.
- It would be extremely rare for a random polypeptide sequence to reliably chelate with zinc in a free ocean. Seawater has a concentration of only about 30 ppb zinc43 but thousands of times greater (about 75 ppm) in the earth’s crust.43 Others report an even lower concentration in seawater, such as 0.6–5 ppb, and rivers containing between 5 and 10 ppb zinc.44 The higher concentration in rivers reinforces the fact that the source of seawater zinc was erosion of the crust, and supposedly three or four billion years ago the concentration of zinc in a primitive ocean would have been far lower still. In addition, zinc is normally not chemically freely available to chelate with polypeptides, but is found firmly associated with other base metals such as copper and lead in ores, and binds well with sulphides instead.41,45
If metal chelation with random polypeptides had played a key role in the origin of proteins, then it would be reasonable for metals such as aluminium and iron, on average about a thousand times more abundant on Earth46 than zinc, to dominate protein chemistry. By similar reasoning, silicon would be expected to play a far more important role than phosphorous, being three hundred times more abundant,46 but the opposite is true.
In a standard reference book on proteins we read,
“Surprisingly elements such as aluminium and silicon that are very abundant in the Earth’s crust (8.1 and 25.7 percent by weight, respectively) do not occur in high concentration within cells. Aluminium is rarely, if ever, found as part of proteins whilst the role of silicon is confined to biomineralization where it is the core component of shells.”47
This is the opposite of what a naturalistic origin for proteins predicts. - The average biological protein is four times larger than 80 AA, posing some difficulties.
- A 2006 study estimated that about 10% of human proteins (2,800) potentially bind zinc.41 Few, if any, biological proteins are composed of domains, with about 80 AA of them all containing zinc. The 10–11 proportion proposed for stable folding was based on a very small domain, and we need to extrapolate to a reasonable-size protein by using the geometric mean of the possibilities. For example, if in the presence of zinc, 10–11 sequences were to produce a native-like fold, but without it the probability were only 10–15, then the probability for a full protein with four domains might be more like [(10–11) x (10–15) x (10–15) x (10–15)]1/4 = 1.0 x 10–14 per domain, or (1.0 x 10–14)4 = 1 x 10–56 for the whole protein.
- Claiming that life could have initiated with much smaller proteins, like 80 AAs, than observed throughout nature today probably does not simplify matters for an evolutionist. Proteins must interact with several other biochemicals, including other proteins, to be able to perform useful processes. If a few hundred multifunctional proteins (of size 300 AA on average) are necessary for autonomous life,48 this number must be several times greater if the numerous functions per protein need to be handled by multiple simpler versions. Furthermore, the functional regions of extant proteins are held together in suitable geometries, facilitating interaction with the multiple intended partners. This would not be true initially for simpler disconnected proteins.

We conclude that the proteins studied do not offer evidence of native-like folding and that the claim that one out of about 1011 random sequences 80 AA long form native-like folds is not well supported.

|

|

Materials and methods used in these studies
Puromycin (figure 10) is an antibiotic that mimics the aminoacyl end of tRNA and terminates translation of mRNA. Figure 11 shows how codons are used by a ribosome to identify the next amino acid to be added to a growing protein. During the last step the protein is located at the P site of the ribosome, and at that time puromycin enters the A site instead of the tRNA which corresponds to the next codon.
To further clarify, the chain-growing reaction which forms a new peptide bond between the amino acids at the A and P sites is shown in figure 12. In nature, puromycin interferes at the A site by forming a stable amide linkage to the nascent protein using its free amino group.
In the methodology developed to create ‘mRNA display proteins’,52 the puromycin is covalently attached to the mRNA of interest and remains so after the mRNA and its newly translated protein is released from the ribosome (figure 13). In this manner the mRNA and resulting protein remain physically linked.
Therefore, isolated proteins which bind to ATP in a column also provide their corresponding mRNA. Copies of those mRNAs can be made using PCR and they can also be deliberately mutated to generate similar variants in the hope of finding better proteins.
References
- For example, the average size of a globular domain according to the CATH database is 153 residues according to: Shen, M-y., Davis, F.P. and Sali, A., The optimal size of a globular protein domain: A simple sphere-packing model, Chemical Physics Letters 405:224–228, 2005. Return to text.
- Lo Surdo, P., Walsh, M.A. and Sollazzo, M., A novel ADP- and zinc-binding fold from function-directed in vitro evolution, Nat. Struct. Mol. Biol. 11:382–383, 2004. See p. 383. Return to text.
- Cho, G., Keefe, A.D., Liu, R., Wilson, D.S. and Szostak, J.W., Constructing High Complexity Synthetic Libraries of Long ORFs Using in Vitro Selection, J. Mol. Biol. 297:309–319, 2000. Return to text.
- Smith, M.D., Rosenow, M.A., Wang, M., Allen, J.P., Szostak, J.W. and Chaput, J.C., Structural Insights into the Evolution of a Non-Biological Protein: Importance of Surface Residues in Protein Fold Optimization, PLoS ONE 2(5):e467, 2007. doi:10.1371/journal.polne.0000467. Pointed out in p. e467. Return to text.
- Axe, D.D., Estimating the Prevalence of Protein Sequences Adopting Functional Enzyme Folds, J. Mol. Biol. 341:1295, 2004. Return to text.
- Keefe, A.D. and Szostak J.W., Functional proteins from a random-sequence library, Nature 410:715–718, 2001. Return to text.
- Keefe and Szostak, ref. 6, p. 715. Return to text.
- en.wikipedia.org/wiki/T7_RNA_polymerase.
T7 RNA Polymerase is an RNA polymerase that catalyzes the formation of RNA in the 5´→ 3´ direction. T7 polymerase is extremely promoter-specific and only transcribes bacteriophage T7 DNA or DNA cloned downstream of a T7 promoter. Return to text. - en.wikipedia.org/wiki/FLAG-tag. The tag contains eight amino acids: DYKDDDDK, with the left-hand end towards the N-terminal and the right-hand end, the C-end of the protein. Return to text.
- en.wikipedia.org/wiki/Polyhistidine-tag. Return to text.
- Cho et al., ref. 3, p. 310. Return to text.
- Keefe and Szostak, ref. 6, p. 716. Return to text.
- CTP: cytidine triphosphate. For the structure, consult any biochemistry or cell biology textbook. Return to text.
- GTP: guanosine triphosphate. For the structure, consult any biochemistry or cell biology textbook. Return to text.
- UTP: uridine triphosphate, For the structure, consult any biochemistry or cell biology textbook. Return to text.
- ITP: inosine triphosphate. For the structure, consult any biochemistry or cell biology textbook. Return to text.
- Keefe and Szostak, ref. 6, p. 717. Return to text.
- CATH domains were downloaded from www.cathdb.info/ domain/1vpcA00. (Version 3.3.0 status 24 June 2009). Entries analyzed with Microsoft Excel. Return to text.
- Simmons, C.R., Stomel, J.M., McConnell, M.D., Smith, D.A., Watkins, J.L., Allen, J.P. and Chaput, J.C., A Synthetic Protein Selected for Ligand Binding Affinity Mediates ATP Hydrolysis, ACS Chemical Biology 4(8):649–659, 2009. Return to text.
- Krishna, S.S. and Grishin, N.B., Structurally Analogous Proteins Do Exist! Structure 13:1125–1127, 2004. Return to text.
- www.geneinfinity.org/rastop/. Return to text.
- www.rcsb.org/pdb/explore/images.do?structureId=1UW1. Return to text.
- Wilson, W., Keefe, A.D. and Szostak, J.W., The use of mRNA display to select high-affinity protein-binding peptides, PNAS 98(7):3750–3755, 2001. Return to text.
- en.wikipedia.org/wiki/Streptavidin#cite_note-0.
“Streptavidin is a 60000 dalton protein purified from the bacterium Streptomyces avidinii. Streptavidin homo-tetramers have an extraordinarily high affinity for biotin (also known as vitamin B7). With a dissociation constant (Kd) on the order of ≈10–14 mol/L, the binding of biotin to streptavidin is one of the strongest non-covalent interactions known in nature. Streptavidin is used extensively in molecular biology and bionanotechnology.” Return to text. - upload.wikimedia.org/wikipedia/commons/5/5a/Streptavidin.png. Return to text.
- en.wikipedia.org/wiki/Biotin.
“Biotin is necessary for cell growth, the production of fatty acids, and the metabolism of fats and amino acids. It plays a role in the citric acid cycle, which is the process by which biochemical energy is generated during aerobic respiration. Biotin not only assists in various metabolic reactions but also helps to transfer carbon dioxide. Biotin may also be helpful in maintaining a steady blood sugar level.
Biotin deficiency is rare, as, in general, intestinal bacteria produce an excess of the body’s recommended daily requirement.” Return to text. - H = Histidine; P = Proline; Q = Glutamine. Return to text.
- Mansy, S.S., Zhang, J., Kümmerle,R., Nilsson, M., Chou, J.J., Szostak, J.W. and Chaput, J.C., Structure and Evolutionary Analysis of a Non-biological ATP-binding Protein, J. Mol. Biol. 371:501–513, 2007. Return to text.
- Mansy et al., ref. 28, p. 502. Return to text.
- Mansy et al., ref. 28, p. 504. Return to text.
- Lo Surdo, ref. 2, p. 382. Return to text.
- As determined from the comments included in the structure data file, www.rcsb.org/pdb/explore/images.do?structureId=1UW1. Return to text.
- www.cathdb.info/cathnode/3. Return to text.
- Garbuzynskiy, S.O., Melnik, B.S., Lobanov, M.Y., Finkelstein, A.V. and Galzitskaya, O.V., Comparison of X-ray and NMR structures: is there a systematic difference in residue contacts between X-ray and NMR-resolved protein structure, Proteins: Struct. Funct. Genet. 60:139–147, 2005. Return to text.
- en.wikipedia.org/wiki/Zinc_finger. Return to text.
- Whitford, D., Proteins: Structure and Function, John Wiley & Sons, Hoboken, NJ, p. 262, 2008. Return to text.
- Wilson et al., ref. 23, p. 3754. Return to text.
- Wilson et al., ref. 23, p. 3755: “Structures of HPQ-containing peptides bound to streptavidin have been determined by x-ray crystallography. The HPQ motif inserts itself into the biotin-binding cleft and forms hydrogen bonds and hydrophobic interactions with streptavidin.” Return to text.
- Chaput, J.C. and Szostak, J.W., Evolutionary Optimization of a Nonbiological ATP Binding Protein for Improved Folding Stability, Chemistry & Biology 11:865–874, 2004. Return to text.
- Smith et al., ref. 4, p. 2. Return to text.
- en.wikipedia.org/wiki/Zinc. Return to text.
- Whitford, ref. 36, p. 25. Return to text.
- Emsley, J., ‘Zinc’, Nature’s Building Blocks: An A–Z Guide to the Elements, Oxford University Press, Oxford, pp. 499–505, 2001. Return to text.
- www.lenntech.com/periodic/water/zinc/zinc-and-water.htm. Return to text.
- Fukai, R. and Huynh-Ngog, L., Chemical Forms of Zinc in Sea Water, J. Oceanographical Society of Japan, 31:179–191, 1975. Return to text.
- en.wikipedia.org/wiki/Abundance_of_elements_in_Earth%27s_crust. Return to text.
- Whitford, ref. 36, p. 7. Return to text.
- Gil, R., Sabater-Muñoz, B., Latorre, A., Silva, F.J. and Moya, A., Extreme genome reduction in Buchnera spp.: Toward the minimal genome needed for symbiotic life, PNAS 99(7):4454–4458, 2002. Return to text.
- en.wikipedia.org/wiki/Puromycin. “Puromycin is an aminonucleoside antibiotic, derived from the Streptomyces alboniger bacterium, that causes premature chain termination during translation taking place in the ribosome. Part of the molecule resembles the 3´ end of the aminoacylated tRNA. It enters the A site and transfers to the growing chain, causing premature chain release. The exact mechanism of action is unknown at this time, but, the 3´ position contains an amide linkage instead of the normal ester linkage of tRNA, the amide bond makes the molecule much more resistant to hydrolysis and thus causes the ribosome to become stopped.” Return to text.
- Wolfe, S.L., Introduction to Cell and Molecular Biology, Wadsworth Publishing Company, Belmont, California, p. 466, 1995. Return to text.
- Wolfe, ref. 50, p. 467. Return to text.
- Roberts, R.W. and Szostak, J.W., RNA-peptide fusions for the in vitro selection of peptides and proteins, Proc. Natl. Acad. Sci. USA 94:12,297, 1997. Return to text.
- Roberts and Szostak, ref. 52, p. 12,301. Return to text.
- Roberts and Szostak, ref. 52, p. 12,300. Return to text.
Readers’ comments
Comments are automatically closed 14 days after publication.