

Journal of Creation 26(1):86–93, April 2012
Browse our latest digital issue Subscribe
The proportion of polypeptide chains which generate native folds—part 6: extraction from random sequences
A series of experiments designed to estimate the proportion of random polypeptides displaying minimal protein folding are evaluated. Candidates were isolated through binding to ATP, exposure to a denaturant, rounds of in vitro selection and error-prone PCR amplification, followed by removal of regions which might interfere with folding. We conclude that the results from circular dichroism spectras and irreversible melts deny the presence of native-like folding in solution under ambient conditions, but rather existence of an artificial zinc-nucleated structure lacking canonical alpha helices or beta strands. There is no reason to believe the polypeptides identified could serve as an initial evolutionary point towards biological proteins with native-like folds.
We point out that although the true proportion of minimally folded proteins among random polypeptides about 300 amino acids in length remains unknown, the current experiments indicate the upper limit must be considerably less than one out of 1011.
In the fifth part of this series on estimates of random polypeptides which will produce native-like folds, we examined1 some experiments initiated at Harvard University by Professor Szostak, winner of the 2009 Nobel Prize in Physiology or Medicine. To our knowledge, only these experiments have attempted to determine experimentally and quantitatively the proportion of random proteins which fold, beginning with random sequences built from the twenty natural amino acids. This work merits careful analysis.
Other studies2–12 have attempted to generate de novo proteins based on rational designs to restrict the size of the search space. These have not permitted a quantitative estimate of the proportion among random protein sequences.
Study number three
The first two series of studies were presented in part 5 of this series1 and the first one was re-examined13 by lead author and Nobel Prize winner Szostak due to some details which had not been communicated before:
“Unfortunately, biophysical characterization of the selected ATP binding proteins proved impossible due to poor solubility.”14
Although the conclusion that about one out of 1011 random polypeptides would produce a stable fold had been disseminated through the earlier publication,15 in this later paper the authors admit that poor solubility, not mentioned before, is a significant detail:
“This observation led to the question of whether protein sequences isolated from unconstrained random sequence libraries could be evolved to adopt a folded state of reasonable stability or whether most such proteins might represent examples of evolutionary dead ends.”13
This is a very good insight. Mutating polypeptide sequences with some degree of thermodynamic stability may often lead to local energy minima and not a true protein native-like fold. Figure 1 illustrates how the starting point must be reasonably close to the sequence of a native-fold if it is to serve as an evolutionary starting point. Such sequences are represented as the top peaks in the fitness landscapes in figure 1. Proteins with insufficient stability and other requirements would be figuratively ‘under water’ and not visible to natural selection. And irrelevant candidates could be viewed as isolated from the mountains, separated by water.
The experimental details12 were explained in part 5 of this series1 and won’t be repeated here. A new experiment was designed13 to isolate more soluble proteins by systematically selecting those which bind to ATP in the presence of a denaturant (GuHCl), which would facilitate opening into a more linear polymer. Clone 18–19 in the earlier work had been found to be sensitive to denaturation (50% loss of ATP binding in 1 M urea),14 which suggests that a stable, native-like fold had not been formed.
The output from round 17 in the study reported earlier1,15 was now used to create a pool of mRNA-displayed proteins, by performing six rounds of in vitro selection and amplification, without deliberately creating additional mutational variants.14
The mRNA-displayed proteins were incubated with ATP-agarose beads in the presence of GuHCl, and the denaturant concentration was increased from 1.5 to 3 M to ensure that less than 10% of the input from each round would remain attached to the ATP-agarose beads, which became the starting point for the next round of selection. In the absence of denaturant (GuHCl), 67% of the material after round six bound to the ATP column, compared to about 35% for the starting sequences in round one.14
Ten sequences from the output of round six were analyzed. Of these, six remained soluble even in the absence of ATP.18 These six were examined in more detail after cloning as maltose-binding protein (MBP) fusion proteins with a thrombin-cleavable linker which permits removal of the MBP portion. These six proteins did not aggregate as easily as the sequences isolated from the earlier work,1,15 during which only binding to ATP had been optimized with no consideration to ensuring they would remain soluble. From the six most soluble sequences, the one binding best to ATP (i.e. having the lowest dissociation constant) labelled B6 was selected for detailed studies. Unfortunately, gel filtration analysis showed that 35% of this material formed undesirable higher-order aggregates.19
Portions of the protein chain were removed systematically by the authors from sequence B6, while ensuring that the two CXXC (Cystein-X-X-Cystein, where X is any residue) portions necessary for chelation with zinc remained intact. The intention was “to eliminate deleterious regions that might nucleate aggregation.”19 This effort was successful, since deletion of the residues at position 74–82 resulted in a protein that was over 98% monomeric according to gel filtration analysis.19 Furthermore, this sequence led to a two-fold improvement in binding to ATP.
Further deletions led to a 62-amino-acid-core ATP binding sequence, labelled B6-62, with comparable affinity for ATP and an elution profile consistent with monomeric protein.
Analogous to the results from the earlier work,1,15 the MBP:B6-62 fusion protein has almost no affinity for ATP analogs CTP, GTP, UTP, ITP and 2-chloro-ATP. Sequential removal of gamma, beta and alpha-phosphate groups in ATP successively raised the dissociation constant,20 and so did modification of the 2’- or 3’-deoxy portions of the ring. These observations led the authors to believe the artificial protein was generating a folded structure amenable to interaction with ATP but not to other substances.
Species B6-62 was further analyzed. Shifts in tryptophan fluorescence in the presence of increasing concentrations of denaturing GuHCl, with and without ATP, “showed a sigmoidal curve consistent with a single cooperative transition between the native and denatured states”.20 “This corresponds to an overall stabilization by 1 nM ATP of 2.7 Kcal/mol.”21
Proton NMR spectra was not consistent with a random coil conformation, and implied some kind of ordering of the protein. The HSQC spectrum22 revealed about 45 well-resolved peaks, which the authors considered consistent with a native-like structure. Unlike clone 18–19, the folding-optimized version seems to be monomeric.23
However, the results from circular dichroism spectroscopy (CD) were completely unexpected! There was no evidence for the secondary structure features (alpha helices and beta sheets) used to form all natural, native-like protein folds.
Analysis of study number three
Is B6-62 truly representative of native-like proteins? The authors provide the same conclusion we reached:
“Our initial data already suggest that this protein has an unusual metal nucleated structure lacking canonical alpha helices or beta strands.”24
In a later paper the thermal stability of the folding-optimized protein and several variants were monitored by changes in circular dichorism. They report that
“Although each of the proteins examined gave sigmoidal curves consistent with a single transition between the native and denatured states, none of the melts were reversible.”25
The CD spectra of clone B6-62 provides the decisive answer:
“This spectral signature is not consistent with standard alpha-helix (two negative bands near 222 nm and 208 nm and a strong positive band near 190 nm), beta-sheet (negative band near 217 nm and positive band near 195 nm), or random coil (strong negative band at 200 nm). One possible explanation for this unexpected CD spectrum is that the structural architecture of this protein is defined by loops collapsed around a zinc-nucleated core. CD spectra taken in the presence of increasing concentrations of ATP did not affect the magnitude or appearance of the 226 nm signal. In contrast, the CD spectrum of B6-62 denatured with 4 M GuHCl changed dramatically, becoming consistent with a random coil peptide, as expected for an unfolded protein.”21
The CXXCXnCXXC zinc-binding site, without which nucleation cannot occur, is not used by any biological proteins known which bind nucleotides.26 This renders the relevance of the studies discussed here of questionable use in evaluating the probability of biological proteins having arisen by chance. It is not surprising that a molecule like ATP offers many binding interaction possibilities with polypeptides, via numerous H-bonds possibilities, or by sandwiching the adenine nucleobase between two protein aromatic side-chains26 in a stable pi-stacking interaction. The phosphate groups could coordinate with any number of polar side-chains.
One must not forget where these improved sequences came from.15 Clone B6 was not present in the original library of 6 x 1012 random sequences. After eight rounds of selecting the mRNA-displayed proteins which bind most strongly to ATP, three consecutive rounds of mutagenic PCR amplification with an average mutagenic rate of 3.7% per amino acid for each round had been carried out.
Recall that B6 had to be judiciously modified to eliminate interfering portions of the chain and to create a soluble variant. In arriving at the denaturing-resistant optimized version B6-62, twelve amino acids along the 62 residue sequence had to be substituted. Of the 2062 = 5 × 1080 possible sequences of 62 residues, a miniscule proportion would have this folding strength. Since the larger 80 AA included portions interfering with folding, it is difficult to justify the claim that 10–11 would be able to fold properly,27 meaning in a manner relevant for biological purposes. And it is far more difficult to imply that this proportion is representative of all proteins, which on average are several times larger than 80 AA.

Study number four
In follow-up studies28,29 a few years after the work summarized in study number one,1,15 clone 18–19 was further optimized for improved folding stability. The pool of protein 18–19 was amplified serially to generate more variants, with error-prone PCR and a target mutagenesis rate of 3.5% per amino acid position.30 The experimental methods match those described in study number three, above, in this paper. Selection for folded variants was performed in the presence of a denaturant (GuHCl) on an ATP-derivatized affinity resin. After five rounds the amount of protein which bound to ATP agarose beads had levelled off to about 40%.30 However, one would expect about 100% of biological ATP-binding enzymes to remain bound.
This work was motivated by the wish to show suitable proteins could arise naturally without being generated by the genetic code. The first sentence of the paper states,
“Primordial enzymes presumably evolved from pools of random sequences, but it is not known how these molecules achieved catalytic function.”31
We read further down the page, “over 3 billion years ago when primordial proteins first appeared on the primitive Earth”. These and other authors repeat elsewhere, “Presumably the first proteins arose from pools of random sequences.”32 Given the difficulties in the RNA World Hypothesis,33,34 it is not surprising an alternative model, that the necessary proteins self-assembled before the genetic code became available, remains popular.
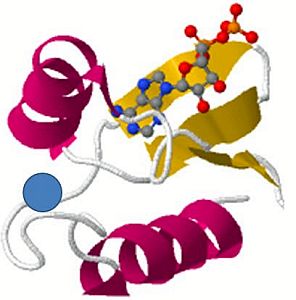
The optimized version of clone 18–19 was called ‘DX’ (double mutant protein) and differed from the progenitor by two amino acids.35 The structure, when crystallized, is shown in figure 2 and includes ADP. The crystal structure reported by two research groups28,36 for 18–19 were essentially identical and also included ADP (figure 3). This was a surprise, since ATP, and not ADP, had been present with the protein during the crystallization process (ADP is ATP with a phosphate group removed).
The authors deduced that in the crystal state the γ-phosphate of ATP forms a strong intramolecular hydrogen bond with the 2’-OH on the sugar ring, lowering the energy of activation to hydrolyze ATP to ADP. The crystal structure of DX suggested that the γ-phosphate of ATP is stabilized with hydrogen bonds to three amino acid side chains40 (Tyr43, Lys34 and Arg41), held in a position which facilitated hydrolysis to ADP. To test this theory, a variant labelled Y43F was created by mutating the tyrosine at position 43 to phenylalanine, which led to a crystal structure which did not hydrolyze ATP (the coordinates are deposited in the protein database under identifier 3DGO). The structure was identical to 3DGL (figure 2).
Unlike in the crystal state, an experiment with thin layer chromatography (TLC) in solution revealed most of the ATP remained bound to the protein and was not hydrolyzed. This is reminiscent of the contradictory information mentioned above: although as a crystal structure, secondary structures seemed to be present in clone B6-62, in solution under ambient aqueous conditions, CD spectra showed these were absent.
Analysis of study number four
However interesting the chemistry reported may be, the structures of biological proteins which catalyze hydrolysis of ATP in a bent position are very different, requiring participation of divalent metal ions and numerous hydrophobic and electrostatic contacts to constrain ATP in a bent geometry.40 Any relevance of the experiments just discussed to biological proteins is only speculation.
Once again, no evidence was found for native-like folding in solution under ambient conditions. A zinc atom chelated with portions of a polypeptide introduces some conformational constraints. Further moulding of a portion of the protein was performed by ATP. The careful laboratory process of creating the crystal, at a very low temperature, caused the artificial protein, already constrained by the zinc, to assume some secondary structure. But these conditions are not expected to be found under natural conditions relevant for evolutionary scenarios.
We believe that the so-called folding behaviour is an artefact, in agreement with what the authors also wrote:
“One interpretation of this result is that, in solution, ATP is able to adopt multiple conformations when bound to protein DX, one of which is the bent conformation whose lifetime is short relative to the time required for hydrolysis. During protein crystallization, the equilibrium between the different bound states shifts to favour the bent conformation, which is observed in the crystal structure of protein DX obtained in the presence of saturating amounts of ATP.”41
However, biological enzymes fold into stable structures under ambient conditions, providing optimal geometric and electronics regions to which their ligands bind reliably.
It is possible that the helices are too short to remain in a single, stable conformation. In one of the leading textbooks on protein chemistry, we read,
“In the α helix, the first four NH groups and last four CO groups will normally lack backbone hydrogen bonds. For this reason very short helices often have distorted conformations and form alternative hydrogen bond partners.”42
The authors interpreted these results as a possible model for primitive enzymes which could eventually evolve into the sophisticated versions found in extant organisms. The opposite conclusion seems more reasonable: it illustrates how valuable ATP, essentially impossible to form under natural conditions on its own, could be indiscriminately destroyed. The useful energy obtained from the hydrolysis of ATP in cells must be tightly regulated and coupled to biochemical networks to harvest this energy.
The authors did not report any data which would permit a quantitative estimate of the random sampling space covered by their directed evolution, which would be many orders of magnitude greater than the original library of 6 x 1012 members.
It is not clear why they conclude that
“… the de novo evolution of this ATP binding protein demonstrates that folded proteins with desired functions occur more frequently than previously thought.”41
On the same page they had just written,
“Since the bent conformation is believed to be essential for hydrolysis, it is possible that this reaction is limited to the crystalline environment where the bent conformation is stabilized by a network of well-ordered water molecules.”
The reader has no way of knowing what proportion of random sequences in a natural setting would create the kind of structure reported for the crystalline state. The latter requires a multitude of identical proteins to be brought together and thermodynamically equilibrated by slow cooling.
Evaluation of studies three and four
The question we are interested in is the proportion of random proteins which would fold into native-like folds, which would then provide a stable scaffold and the potential to be useful for some cellular function. No-one questions that ATP, and other large organic molecules, can interact with a variety of substances, including polypeptides and even amorphous tar-like material.
Of fundamental importance in evaluating these studies is to be careful in how the term ‘protein fold’ is used. Both the CATH43 and SCOP44 protein classification systems use the technical term ‘fold’ to define precise three-dimensional topologies, and the reader could be misled to believe a true fold has been identified under ambient conditions for the synthetic proteins reported.
Intended interactions of biological proteins with ATP are strong and very selective, such as binding of the adenine ring by protein kinases.45 Inspection of the chemical structure of ATP1 reveals many locations where accidental non-covalent interactions with polypeptides can occur. Hydrogen bonds can form with the amino, hydroxyl and phosphate groups and so can Van der Waals interaction with the flat, aromatic adenine ring. Therefore, it is not surprising that about 0.1% of the random proteins generated were reported to be able to bind to ATP in the seminal paper.46 Eight rounds of selection, without creating additional variety, increased the fraction which binds to 6.2%. This only demonstrates that some portions of the protein can interact with the much smaller ATP molecule, an observation chemically expected.
Some criteria to characterize native-like folded proteins
We can contrast the characteristics of the polypeptides isolated with those of typical folded proteins:
- Presence of secondary structures features, such as alpha helices, beta sheets and stabilized connecting coils. Typically more than 60% of a protein folds into alpha helices and beta sheets.47
- Self-assembly of a single three-dimensional topology, before interaction with other bio-chemicals to form the quaternary structure. The amino acid sequence, and not structural information obtained from some template, determines its folding.48,49 If denatured non-destructively, such as by using organic solvents (e.g. urea) or detergents (e.g. sodiumdodecyl sulphate), they can usually refold quickly back to the native state once the denaturant is removed. No cofactors are necessary for the self-assembly,48 initially nor after removing the non-destructive50 denaturing reagents by dialysis.
- Folding into a single, lowest-energy state. This also takes into account constraints to funnel the folding process into the correct topology by using sequence details which generate impediments to folding into an incorrect local energy minimum.
- Solubility in water.51
- Formation of a discrete protein species, usually monomeric. High-order aggregates often result from misfolded proteins52–54 and underlie many diseases. This implies that ‘sticky’ hydrophobic patches on the surface must be avoided. Higher order aggregates must use precise and reliable locations for interactions, and not lead to a chaotic mixture of different aggregation states nor interaction sites.
Researchers believe that folding often involves four steps:55
- The unfolded stage, which actually lacks truly randomised structure.56 This is followed by …
- Formation of secondary structures. These first elements of secondary structure are only metastable, but it is believed that when enough interact by random movements, a cumulative stabilizing effect can arise, leading to more stable collections of secondary structure.57 Soluble proteins are driven by hydrophobic interactions that force non-polar residues together in the central core of the protein. This condensation is called a ‘hydrophobic collapse’.58 This leads to …
- The molten globule which is more compact and resembles the native protein in overall shape, but lacks many of the non-covalent side-chain bonds, covalent disulfide bonds, charge pair interactions and other stabilizing features which characterize the final state. Molten globules are still quite open and flexible, and the secondary structures may not be entirely formed.59
- The final state is the native-like fold.
Some proteins do not appear to go through a molten globule intermediate. But more significantly, in vitro studies reveal the presence of additional intermediates which are not along the pathway towards a native state. In vivo specific proteins catalyse the elimination of ‘unwanted’ folds.60 The existence of other conformational states, which partially share characteristics with truly native folds, is probably the key to understanding the structure of clone 18–19.
Interpretation of the experimental data
Interaction of several AAs with a ligand does not automatically imply a native protein fold is present. All biological native-like folds are composed of alpha helices and/or beta strands, unlike the structure of the reported proteins, such as the optimized B6-62:
“Given the unusual CD spectra indicating the absence of significant alpha helix or beta strand contribution we decided to investigate ….”61
Now, circular dichroism (CD) in the near and far UV range shows different spectra for native, folded and molten globule states.65 It is also used routinely to identify alpha helices, beta structure motifs, and random coils.62 We agree with the author’s interpretation of the clone B6-62:
“However, the results of CD spectroscopy suggest that the folded protein has an unusual structure that may differ from normal biological protein structures.”63
Earlier we proposed1 that the minimal secondary structure suggested by crystallographic analysis of clone 18–19 was probably an artefact and not representative of the state under ambient aqueous conditions. Crystallographers are aware64 that the process of crystallization of pure substances to permit X-ray diffraction can generate a conformation very different from that found under ambient conditions. For proteins this is especially problematic, due to the only marginal stability of the best conformation:
“Although helices have regular repeating hydrogen bonds coupled with a uniformity of bond lengths and angles this periodicity masks their marginal stability. In an isolated state most helices will unfold”.65
Stabilization through chelation with zinc would create small constrained regions on polypeptides without requiring the kinds of secondary structures found in biological proteins. If such a structure would remain stable, portions could interact with a variety of organic substances. To illustrate, a tiny portion of antibodies (which are proteins) are highly variable, permitting interaction with millions of different ligands (antigens).66,67 However,
“Only 5–10 amino acids in each hypervariable region form the antigen-binding site. As a result, the size of the antigenic determinant that an antibody recognizes is generally comparably small. It can consist of fewer than 25 amino acids on the surface of a globular protein, for example.”68
In a prebiotic world with no genetic code producing many identical proteins, naturalistic scenarios must take into account the great variety of different substances which would be present, among which useful protein sequences are supposed to be found. The evidence we have examined illustrates that many worthless and deleterious chemical interactions can occur. As an example,69 molecules like ATP can also bind to random DNA sequences, in addition to random proteins, in a fairly high proportion.
Evolutionary scenarios must take into account the need to prevent deleterious chemical interactions. As pointed out earlier,1,70 clone 18–19 hydrolyzes ATP, thereby destroying one of the most important molecules used by cells, in an unregulated manner which would serve no purpose.
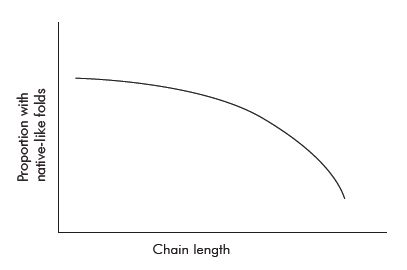
B6-62 is almost three times smaller than an average domain.73 This was formed after the authors judiciously removed portions of protein B6 to avoid aggregation and insolubility problems. The proportion of native-like folded proteins decreases as the chain length increases. No plausible native-live candidates were identified from the initial library of 6 × 1012 sequences, from which the best candidates were amplified and mutated to enhance the chance of finding a suitable variant. Therefore, the limited data suggests that the proportion of native-like folded domain in a random library could well be less than 10–11 and many orders of magnitude lower for proteins which consist of multiple domains (which the majority do).
Besides the rapidly increasing tendency to aggregate and become insoluble with increasing chain length (figure 4), there are other reasons why larger, biologically relevant proteins of average size 300 AA are more difficult to create using natural processes.
One example is that proline residues (figure 5) need to isomerize to the correct isomer. The cis and trans isomers of the X-Pro peptide bond (where X represents any amino acid) are nearly equal energetically. The fraction of X-Pro peptide bonds in the cis isomer form when unconstrained typically ranges from 10–40%. Cis–trans proline isomerization is a very slow process that can prevent correct protein folding by trapping one or more proline residues crucial for folding in the non-native isomer, especially when the native protein requires the cis isomer.71,72 Clone 18–19 has only three prolines, but an average-size domain of 150 AA73 would have about 10 prolines, given that 4/61 codons code for proline, and an average-size protein of 300 AA would have 20 prolines.

All organisms have prolyl isomerase enzymes to catalyze this isomerization72,74 but in a prebiotic scenario the native folds would often not be produced in a relevant time period.
As another example, the number of disulphide bonds increases with protein size and these could form indiscriminately, hindering or preventing formation of the native state in a pre-biotic scenario.
Conclusion
We cannot agree that this series of experiments demonstrates that about 10–11 random protein sequences produce native-like folds. The estimate that 10–11 random polypeptides would possess a native fold was based17 on clone 18–19. Additional information surfaced after this proposed estimate had been widely disseminated. Recall that the folding results were only possible in the presence of zinc, that “protein 18–19 forms an insoluble precipitate after 3 days”75 and that high concentrations of ATP were necessary for it to remain stably folded.30 Surely clone 18–19 cannot be considered a representative evolutionary starting point for a new true protein.
The DX variant with better solubility was not part of the original 1013 random sequences but of a larger space of random sequences, and therefore the proportion which folds properly needs to be calibrated accordingly. That the value is lower than 10–11 is certain, but how much lower? The secondary structures reported were found in crystals frozen76 at 90 K but were not found in the CD spectra at evolutionary-relevant temperatures. DX is also contingent upon the presence of chelating zinc. Furthermore, ATP was also found77 associated with a β-sheet in the crystal structure of DX, suggesting that all or most secondary structure could be an artefact, caused by zinc and ATP, forming only at temperatures around 100 K (–173°C).
Final comments
In Part 1 of this series,78 I tried to find a lower limit for protein folding everyone could agree with. I pointed out that all combinations of binary patterning79 for helices and coils would be covered by a polypeptide pattern such as:
(p/n)AA(p/n)AA,
where p = polar; n = non-polar; A = Anything. This is a very generous assumption. This implies for an 80-residue domain that only every third position would pose a constraint, leading to a proportion of (9/20)27 = 10–10. Since this is actually an order of magnitude greater than proposed15 in the studies analyzed here, perhaps others would be willing to accept the reasonableness of my lower bound. This implies that a proportion of about (9/20)100 = 10–35 random polypeptides 300 residues long, about an average size protein, would fold properly.
Of great value would be experiments to isolate and characterize properly folded random polypeptides in the absence of ligands and chelating metals, especially those metals expected to be present in extraordinarily low concentration in a primitive ocean.
References
- Truman, R., The proportion of polypeptide chains with native folds— part 5: experimental extraction from random sequences, J. Creation 26(1):76–85, 2012. Return to text.
- DeGrado, W.F., Summa, C.M., Pavone, V., Nastri, F. and Lombardi, A., De novo design and structural characterization of proteins and metalloproteins, Annu. Rev. Biochem. 68:779–819, 1999. Return to text.
- Moffet, D.A. and Hecht, M.H., De novo proteins from combinatorial libraries, Chem. Rev. 101:3191–3203, 2001. Return to text.
- Roy, S., Ratnaswamy, G., Boice, J.A., Fairman, R., McLendon, G. and Hecht, M.H., A protein designed by binary patterning., J. Am. Chem. Soc. 119:5302–5306, 1997. Return to text.
- West, M.W., Wang, W., Patterson, J., Mancias, J.D., Beasley, J.R. and Hecht, M.H., De novo amyloid proteins from designed combinatorial libraries, Proc. Natl. Acad. Sci. USA 96:11211–11216, 1999. Return to text.
- Wei, Y., Kim, S., Fela, D., Baum, J. and Hecht, M.H., Solution structure of a de novo protein from a designed combinatorial library, Proc. Natl. Acad. Sci. USA 100:13270–13273, 2003. Return to text.
- Davidson, A.R. and Sauer, R.T., Folded proteins occur frequently in libraries of random amino acid sequences, Proc. Natl. Acad. Sci. USA 91:2146–2150, 1994. Return to text.
- Davidson, A.R., Lumb, K.J. and Sauer, R.T., Cooperatively folded proteins in random sequence libraries, Nat. Struct. Biol. 2:856–864, 1995. Return to text.
- Desjarlais, J.R. and Handel, T.M., De-novo design of the hydrophobic core of proteins, Protein Sci. 4:2006–2018, 1995. Return to text.
- Dahiyat, B.I. and Mayo, S.L., De novo protein design: fully automated sequence selection, Science 278:82–87, 1997. Return to text.
- Voigt, C.A., Mayo, S.L. Arnold, F.H. and Wang, Z.-G., Computational method to reduce the search space for directed protein evolution, Proc. Natl. Acad. Sci. USA 98:3778–3783, 2001. Return to text.
- Kuhlman, B., Dantas, G., Ireton, G., Varani, G., Stoddard, B.L. and Baker, D., Design of a novel globular protein fold with atomic-level accuracy, Science 302:1364–1368, 2003. Return to text.
- Chaput, J.C. and Szostak, J.W., Evolutionary optimization of a nonbiological ATP binding protein for improved folding stability, Chemistry & Biology 11:865–874, 2004. Return to text.
- Chaput and Szostak, ref. 13, p. 866. Return to text.
- Keefe, A.D. and Szostak J.W., Functional proteins from a random-sequence library, Nature 410(6829):715–718, 2001. Return to text.
- Axe, D., Extreme functional sensitivity to conservative amino acid changes on enzyme exteriors, J. Mol. Biol. 301:585, 2000. Return to text.
- Roberts, R.W. and Szostak, J.W., RNA-peptide fusions for the in vitro selection of peptides and proteins, Proc. Natl. Acad. Sci.USA 94:12297, 1997. Return to text.
- Chaput and Szostak, ref. 13, p. 867. Return to text.
- Chaput and Szostak, ref. 13, p. 869. Return to text.
- Chaput and Szostak, ref. 13, p. 870. Return to text.
- Chaput and Szostak, ref. 13, p. 871. Return to text.
- Heteronuclear single quantum coherence, en.wikipedia.org/wiki/ Heteronuclear_single_quantum_coherence. Return to text.
- Mansy, S.S., Zhang, J., Kümmerle, R., Nilsson, M., Chou, J.J., Szostak, J.W. and Chaput, J.C., Structure and evolutionary analysis of a non-biological ATP-binding protein, J. Mol. Biol. 371:501–513, 2007; see p. 505. Return to text.
- Chaput and Szostak, ref. , p. 872. Return to text.
- Mansy et al., ref. 23, p. 504. Return to text.
- Mansy et al., ref. 23, p. 506. Return to text.
- For example, if 4 of the 20 amino acids had been acceptable for the folding optimized version, the proportions would be (4/20)12 = 4 x 10–9 multiplied by the sequences samples during the first 8 rounds multiplied by the sequences beside the 12 positions which did not lead to improvement. Note also that the folding optimized version differed from sequence 18–19 by 12 amino acids. Return to text.
- Smith, M.D., Rosenow, M.A., Wang, M., Allen, J.P., Szostak, J.W. and Chaput, J.C., Structural insights into the evolution of a non-biological protein: importance of surface residues in protein fold optimization, PLoS ONE 2(5):e467, 2007; doi:10.1371/journal.polne.0000467. Return to text.
- Simmons, C.R., Stomel, J.M., McConnell, M.D., Smith, D.A., Watkins, J.L., Allen, J.P. and Chaput, J.C., A synthetic protein selected for ligand binding affinity mediates ATP hydrolysis, ACS Chemical Biology 4(8):649–659, 2009. Return to text.
- Smith et al., ref. 28, p. 2. Return to text.
- Simmons et al., ref. 289, p. 649. Return to text.
- Cho, G., Keefe, A.D., Liu, R., Wilson, D.S. and Szostak, J.W., Constructing high complexity synthetic libraries of long orfs using in vitro selection, J. Mol. Biol. 297:309–319, 2000; see p. 310. Return to text.
- Gibson, L.J., Did life begin in an ‘RNA world’? Origins 20(1):45–52, 1993; www.grisda.org/origins/20045.htm. Return to text.
- Mills, G.C. and Kenyon, D., The RNA world: a critique, Origins & Design 17:1, 1996; www.arn.org/docs/odesign/od171/rnaworld171.htm. Return to text.
- Simmons et al., ref. 29, p. 650. Return to text.
- Lo Surdo, P., Walsh, M.A. and Sollazzo, M., A novel ADP-and zinc-binding fold from function-directed in vitro evolution, Nat. Struct. Mol. Biol. 11:382–383, 2004. Return to text.
- www.rcsb.org/pdb/explore/explore.do?structureId=3DGL. Return to text.
- www.geneinfinity.org/rastop/. Return to text.
- www.rcsb.org/pdb/explore/images.do?structureId=1UW1. Return to text.
- Simmons et al., ref. 29, p. 652. Return to text.
- Simmons et al., ref. 29, p. 655. Return to text.
- Whitford, D., Proteins: Structure and Function, John Wiley & Sons, West Sussex, England, p. 41, 2008. Return to text.
- www.cathdb.info/. Return to text.
- scop.mrc-lmb.cam.ac.uk/scop/. Return to text.
- Lodish H. et al., Molecular Cell Biology, 4th ed., W.H. Freeman and Company, New York, p. 71, 2000. Return to text.
- Keefe and Szostak, ref. 15, p. 715. Return to text.
- Stryer, L., Biochemistry, 4th ed., W.H. Freeman and Company, New York, p. 423, 1999. Return to text.
- Lodish et al., ref. 45, p. 62. Return to text.
- Whitford, ref. 42, p. 402. Return to text.
- Destructive denaturing could be performed using extreme heat or UV light, modifying the chemical nature of the amino acids. Return to text.
- We are neglecting the small proportion of membrane proteins, which need not be water soluble. Return to text.
- Whitford, ref. 42, p. 427: “The protein aggregates are linked by formation of elongated fibrils (amyloid) and diseases showing this property are collectively grouped together by the term amyloidosis.” Return to text.
- Whitford, ref. 42, p. 431: prions are an example of a misfolded protein which leads to further aggregation. It is the cause of diseases such as scrapie, Creutzfeldt-Jakob disease and Kuru. Return to text.
- Prusiner, S.B, Scott, M.R., DeArmond, S.J. and Cohen, F.E, Prion protein biology, Cell 93:337, 1998. Return to text.
- Karp, G., Cell and Molecular Biology, 2nd ed., John Wiley & Sons, New York, chap. 2.5, 1999. Return to text.
- Whitford, ref. 42, p. 396. Return to text.
- Whitford, ref. 42, p. 410. Return to text.
- Stryer, ref. 47. p. 419. Return to text.
- Alberts, B. et al., Molecular Biology of the Cell, 4th edition, Garland Publising, New York, p. 213, 2002. Return to text.
- Whitford, ref. 42, pp. 412, 415. Return to text.
- Chaput and Szostak, ref. 13, p. 871. Return to text.
- Stryer, ref. 47. p. 62. Return to text.
- Chaput and Szostak, ref. 13, p. 872. Return to text.
- The author was trained in crystallography and had an internship at Brookhaven National Lab in this field. Return to text.
- Whitford, ref. 42, p. 409. Return to text.
- en.wikipedia.org/wiki/Antibody. “This region is known as the hypervariable region. Each of these variants can bind to a different target, known as an antigen. This huge diversity of antibodies allows the immune system to recognize an equally wide variety of antigens. The unique part of the antigen recognized by an antibody is called the epitope. These epitopes bind with their antibody in a highly specific interaction, called induced fit, that allows antibodies to identify and bind only their unique antigen in the midst of the millions of different molecules that make up an organism. Recognition of an antigen by an antibody tags it for attack by other parts of the immune system.” Return to text.
- Alberts et al., ref. 59, p. 1376. “The basic structural unit of an antibody molecule consists of four polypeptide chains, two identical light (L) chains (each containing about 220 amino acids) and two identical heavy (H) chains (each usually containing about 440 amino acids).” Return to text.
- Alberts et al., ref. 59, p. 1382. Return to text.
- Sassanfar, M. and Szostak, J.W., An RNA motif that of the ATP binding protein was solved by X-ray crystallography and binds ATP, Nature 364:550–553, 1993. Return to text.
- www.rcsb.org/pdb/explore/images.do?structureId=1UW1. Return to text.
- en.wikipedia.org/wiki/Proline. Return to text.
- Whitford, ref. 42, p. 414. Return to text.
- The average size of a globular domain according to the CATH database is 153 residues. Shen, M-y., Davis, F.P. and Sali, A., The optimal size of a globular protein domain: A simple sphere-packing model, Chemical Physics Letters 405: 224–228, 2005. Return to text.
- en.wikipedia.org/wiki/Proline. Return to text.
- Smith et al., ref. 28, p. 5. Return to text.
- Smith et al., ref. 28, p. 9. Return to text.
- Smith et al., ref. 28, p. 6. Return to text.
- Truman, R., The proportion of polypeptide chains which generate native folds—part 1: analysis of reduced codon set experiments, J. Creation, 25(1):77–85, 2011. Return to text.
- Kamtekar, S., Schiffer, J.M., Xiong, H., Babik, J.M. and Hecht, M.H., Protein design by binary patterning of polar and nonpolar amino acids, Science 262:1680–1685, 1993. Return to text.
Readers’ comments
Comments are automatically closed 14 days after publication.