
Does Genetics Point to a Single Primal Couple?
A response to claims to the contrary from BioLogos
Jon P., from the United States, wrote in with questions about a recent article on our site titled Adam, Eve and Noah vs. Modern Genetics. He first quotes from the CMI article, then references an article that appeared on the theistic evolutionary BioLogos website that supposedly contradicts it. His short letter is printed in full followed by a response from Robert Carter.
Jon P. writes:“The important take home point is that essentially all of the genetic variation among people today could have been carried within two people,…”
What about what Biologos says that contradicts what you have written? [Link removed as per Feedback rules.]
Dr. Carter responds:
Dear Jon,
I am familiar with Falk and Venema’s article, having read it when it was first posted, and am pleased to finally have an excuse to write a retort.
First, you must read Lita Sanders’s article Evolutionary syncretism: a critique of BioLogos. You will also want to consult A response to Timothy Keller’s ‘Creation, Evolution and Christian laypeople’.
Second, regarding the science, we must ask, “What would we expect the data to indicate if Adam and Eve actually existed?” As I attempted to demonstrate in my article, the data fit very nicely into the biblical model of one founding couple for all of humanity who lived a few thousand years ago. There was one founding female (directly predicted by the creation model and allowed in the evolution model, so this not really evidence for either side). There was one founding male (also non-conclusive). There are three main mitochondrial lineages in the world, found (unequally) among the people groups scattered across the world, with only a few differences between the founder lineages of the three groups (a better fit to the creation model, why did only three sub-lineages of closely-related mtDNAs escape Africa?, etc.). There was a single dispersal of people in the recent past (a better fit to the creation model, but Out of Africa can also be used as an explanation). There was a population bottleneck (a direct prediction of the biblical model and an ad hoc addition to the Out of Africa theory). In short, there are a lot of things in genetics that support the biblical narrative. This would not be expected if Genesis was a random and made-up story (or collection of stories).
You might also be interested in what we have already written on the subject, including the first seven articles (at the time of this writing) on our Genetics Q&A page.
Now, let us deal with the meat of Falk and Venema’s arguments. They give three supposedly independent lines of evidence that point away from Adam and Eve and toward standard evolutionary history. I will treat each in turn:
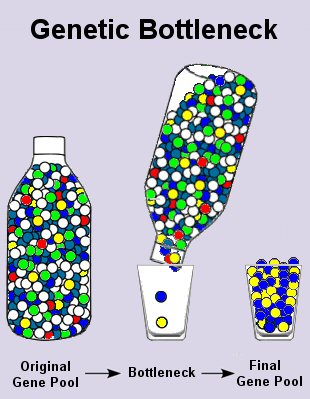
Method 1: Bottleneck
The authors claim 1) there was a dramatic reduction in the size of the human population in the past (this is called a “genetic bottleneck”), but that 2) this does not square with the biblical model of Adam and Eve. They are conflating several things here and seemed confused about what the Bible states about possible bottlenecks. I know what they are trying to do, but there are two separate issues here: the creation of Adam and Eve and the bottleneck that occurred during Noah’s Flood about 1,600 years later.
They say the genetic bottleneck caused by the creation of Adam and Eve would be extreme, since a maximum of only four versions of every genetic variation could be contained with Adam and Eve (because each carried two copies of each chromosome: 2+2=4). What they fail to point out, however, is that most genetic variation comes in two versions and these versions are found distributed throughout the world’s population. When we find more variation than that, the third, fourth, etc., versions of the gene in question are usually restricted to specific subpopulations. In other words, these are mutations that have occurred since the human population spread out after Babel.
Falk and Venema say:
“In short, the genetic impact of such an event would leave a stamp on the genome of that species that would persist for tens of thousands of generations as mutations slowly generated genetic diversity.”
But this is exactly what we are seeing. There is a surprising lack of diversity among people worldwide. Most of the diversity is shared among all populations, however, and diversity is increasing over time due to mutation in each of the sub populations.
They go on to say that there is abundant evidence for a population bottleneck, but not the biblical one. Part of their error comes from the fact that they do not consider the bottleneck that occurred at the Flood, where the entire world’s population was reduced to three reproducing couples. My article discusses this in more depth.
Another part of their error is their appeal to mutation rate theory to calibrate the size of the ancestral population during the bottleneck. The mutation rates used in the calculations generally depend on assumptions of common ancestry,1 which is the very question we are trying to answer. One cannot use an assumption to prove itself! Measurable mutation rates are generally several orders of magnitude faster than those used in evolutionary studies. Using a measured rate would shrink the size of the bottleneck population.
Another part of their error comes from the fact that the authors substitute an evolutionary bottleneck for the biblical one. In evolutionary mathematics and theory, one rarely considers a single-generation bottleneck. Rather, a “bottleneck” is defined as a restriction in the population size that lasts long enough to have an effect. A single-generation event (e.g., Noah’s Flood), with a rapid expansion of the population afterward (prior to and after Babel), will lead to a negligible loss of diversity.
An interesting illustration of this can be seen in Figure 6.2 of Sanford et al. (2007).2 In this example, a 500-generation bottleneck leads to a faster loss of “fitness” than would have been true without the bottleneck. Genetic diversity also goes down, but this is not being shown in the graph. I do not have a convenient reference that illustrates a one-generation bottleneck, but you can reproduce one using the genetic modeling program, Mendel’s Accountant (see reference above). Short bottlenecks have little effect on genetic diversity.
The three founding Ark couples could have carried a huge percentage of the pre-Flood genetic variation (Adam and Eve’s diversity + pre-Flood mutations), with the exception of the Y chromosome (total loss of all but one line) and mtDNA (restricted to three lines). Hence, the biblical model fits the available data nicely and the data do not serve as an easy refutation of Adam and Eve.
Method 2: Alus
Alus are small pieces of DNA that occur all over the human genome. Historically, they have been considered to be “junk DNA”, free to jump around and insert themselves into the genome at will.
The authors assume Alu insertions are random. Thus, they treat Alus in the traditional sense, as a class of ‘junk DNA’. My quote from Methane, Ethane, and Pseudogene Functions is appropriate to restate here: “The junk DNA argument is like a zombie. It is dead and buried, but keeps coming back … Since nothing else is waiting in the wings, they desperately cling to a decaying corpse of a theory.”
Please read The slow, painful death of junk DNA. In this article, I discuss another group of “jumping genes”, retrotransposons, and how our understanding of them has shifted to the point where we now believe they are decidedly functional. I predict Alus are going to fall into the functional class as well. Think about it, they represent a pervasive and active class of DNA elements, with many subfamilies and insertion points unique to humans. They subsume a sizable percentage of the human genome. They are amazingly similar among themselves and among the world population. If they were “junk” they should be slowly mutating to oblivion. And, they have to be maintained. A significant amount of energy and time is taken up by the cell to maintain Alus. Would not selection pressures weed out these parasitic elements over time? The rule in biology is that form follows function. Alus have a strong form, therefore…
As functional genetic elements, Alus probably do not jump randomly. Their argument is based on the assumption of random jumping. If jumping and insertion is predictable due to environment or a pre-programmed genetic algorithm that controls Alu movement, the argument fails. Time will tell who is correct, but, like Method #1, their conclusions are driven by their starting assumptions.
Method 3: HapMap data
The HapMap Project3 was designed to catalog a significant fraction of human genetic diversity. They analyzed millions of variants in thousands of people from around the world and made the data freely available. One might wonder, if the HapMap is so unfriendly to the creation model, why do I have much of that data stored on my hard drive? In fact, I am quite happy with what we learned from the project (I am even happier with the 1000 genomes project, which is a step above HapMap, but which is not yet complete). What have we learned?
- The human genome is young: shared blocks of DNA are large and there has not been enough time to scramble them to randomness.
- The human population came from a single source: most blocks are shared among all world populations.
- The human genome is falling apart: deletions tend to NOT be shared among populations, but are unique to subpopulations (this is further evidence for the youth of the genome and that we came from a single source population in the recent past).
The authors once again assume evolutionary theory in their argument. They also assume recombination is random. If it is not, most of what they say falls apart. But the latest data indicate recombination is controlled by genetic factors that vary among populations and that change over time.4
We have a lot of work yet to do on this, but the data are not contradictory to Adam and Eve being our ancestors and living only 6,000 years ago. In fact, the genetic diversity God put into Adam and Eve is still being scrambled by recombination. I hope that, one day, some enterprising creationist is going to take the data (when enough accumulates) and use it to backtrack human history, arriving at Adam and Eve. For now, however, I am comfortable that there is no obvious disproof of Adam and Eve in the HapMap data, despite the authors’ assertions to the contrary.
Summary:
Falk and Venema claim their three methodologies are independent, but they are not, for each is based on evolutionary assumptions. Not only that, but they are not necessarily rock-solid, even using evolutionary arguments. Also, none of the three “big arguments” falsifies creation. In fact, I am quite at ease with all of them and will happily use them as arguments for creation in the future.
Thanks for a great question and for giving me the opportunity to wax eloquent on one of my favorite subjects.
Sincerely,
Robert Carter
References
- E.g., if chimps and humans have been separated for 6 million years and there are N differences between two genes shared between the two species, the mutation rate would be N/6,000,000. Return to text.
- Sanford, J., et al., Mendel’s Accountant: A biologically realistic forward-time population genetics program. SCPE 8(2):147-165, 2007. This paper can be accessed here: http://mendelsaccount.sourceforge.net/ (scroll down to the references). Return to text.
- HapMap.org Return to text.
- Parvanov, E.D., et al., Prdm9 controls activation of mammalian recombination hotspots. Science 327:835, 2010; Berg, I.L., et al., PRDM9 variation strongly influences recombination hot-spot activity and meiotic instability in humans. Nature Genetics 42(10):859-864, 2010. Return to text.



Readers’ comments
Comments are automatically closed 14 days after publication.