

Journal of Creation 20(2):90–99, August 2006
Browse our latest digital issue Subscribe
Searching for needles in a haystack
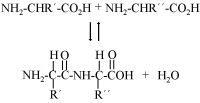
The variability of amino acids in polypeptide chains able to perform diverse cellular functions has been shown in many cases to be surprisingly limited. Some experimental results from the literature are reviewed here. Systematic studies involving chorismate mutase, TEM-1 β lactamase, the lambda repressor, cytochrome c and ubiquitin have been performed in an attempt to quantify the amount of sequence variability permitted. Analysis of these sequence clusters has permitted various authors to calculate what proportion of polypeptide chains of suitable length would include a protein able to provide the function under consideration. Until a biologically minimally functional new protein is coded for by a gene, natural selection cannot begin an evolutionary process of fine-tuning. Natural selection cannot favour sequences with a long term goal in mind, without immediate benefit. An important issue is just how difficult statistically it would be for mutations to provide such initial starting points. The studies and calculations reviewed here assume an origin de novo mainly because no suitable genes of similar sequence seem available for these to have evolved from. If these statistical estimates are accepted, then one can reject evolutionary scenarios which require new proteins to arise from among random gene sequences.
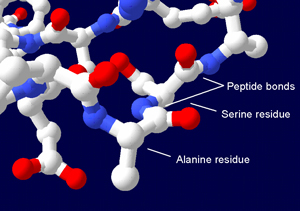
Proteins are chemically bonded chains of amino acids (AAs) (figure 1). All living organisms on Earth depend on specialized services these provide. One of 20 different AAs1 can be placed at each residue position of the polypeptide, offering an immense sequence space of possible variants. Most alternatives are biologically useless.
Many scientists, including several prominent agnostics, are persuaded that Darwinian trial-and-error could not have produced the necessary genetic infrastructure for life to be possible.2 The fraction of all possible AA chains having any biological value is miniscule. Requiring hundreds of unrelated combinations of amino acids forming polypeptides, in the right proportion and same place, for the simplest of autonomous life forms to be possible, is indistinguishable from demanding a miracle. Additional requirements for other classes of biochemicals found in all cells compounds the improbability. The minimal requirements for a putative initial evolutionary starting point via naturalist means cannot be justified from what is known from chemical and thermodynamical principles.
We will limit this discussion to real, biological, genetically based organisms and exclude speculative constructs such as abstract ‘replicators’,3 RNA-world arguments4 and ‘chemical hyper-cycles’.5 Even if such hypothetical structures could exist at some point, a transformation to life as we know it, based on the genetic code, would confront us with the issues discussed here anyway.
For a primitive organism to evolve and increase the range of functions performed, many new kinds of genes are needed. It has been proposed that different genes may have arisen from duplicated copies6 on the same genome, which diverged through mutations and ended up coding for novel proteins. I believe this concept has limited explanatory potential. The number of mutational trials needed to arrive at truly novel genes is prohibitive given the great differences observed among families of unrelated proteins. Nevertheless, divergence of paralogous genes (duplicates on the same genome) and lateral gene transfer remain key explanatory notions in the evolutionary toolkit. This is justified, since we will see here that de novo origin of proteins in living organisms is statistically not plausible. An analysis of duplicate genes and lateral gene transfer will follow in later papers.
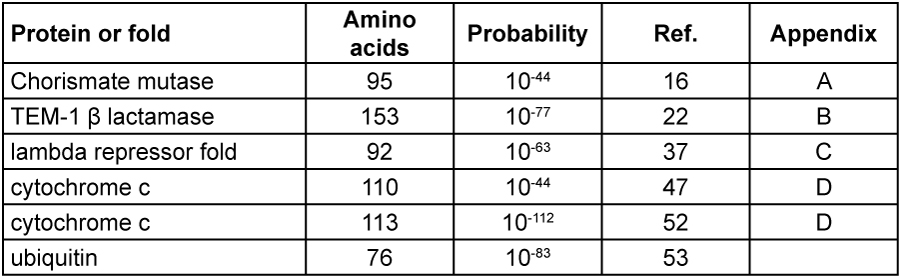
Just how difficult would it be for mutations to generate new valuable genes by chance? It depends for one thing on what fraction of random amino acid chains would provide new useful functions with enough advantage for natural selection to act upon. The conclusions from several published studies have been summarized in Table 1. The technical details are discussed in the Appendices as an aid to those wishing to understand the original literature.
Three general approaches are described in the literature to examine the proportion of sequences able to provide a particular protein function: (1) random chains of amino acids are generated to see if useful variants appear; (2) existing protein sequences are mutated to see how much change is tolerated; (3) sequence variability across organisms is compared. Especially interesting are those cases where no, or few, similar protein classes are available from which the highly restricted version could plausibly have derived. This is an area I and other non-evolutionists are currently researching.
Testing of random polypeptide sequences
In this approach, many polypeptide sequences are randomly generated and tested for some property related to that of functional proteins. This literature7–11 will not be reviewed at this time. I have searched the literature for years without success for an example in which anything useful for the organism was claimed using this approach. Examples of, for example, stability to proteolysis7 or cooperative denaturation,8 even crude catalytic effects,12 are certainly chemically interesting, but these do not yet provide plausible starting points for Darwinian selection to take place. It is important to keep in mind that expressed genes cost considerable energy resources,13 and mere analogy to properties used by real proteins is not something natural selection can act upon.
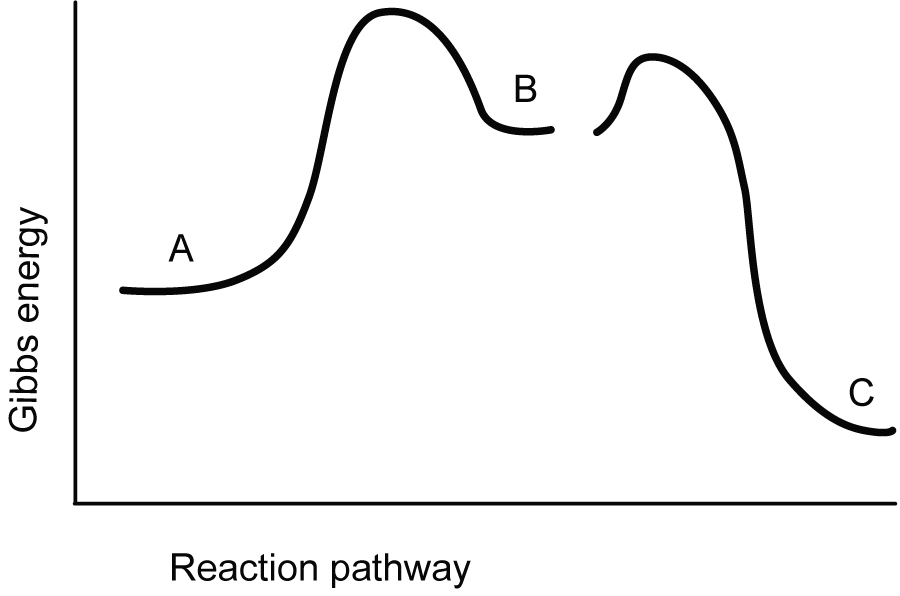
A new gene which produces a polypeptide serving no useful function which is merely harder to degrade, will not provide a selective advantage. In fact, being unable to degrade and recycle such building material in a regulated manner14 would be disadvantageous. Furthermore, it appears that the potential for interference in existing processes would simply be introduced. Crude enzymes accelerate the forward and backward reaction by lowering transition state energies (figure 2), and could simply facilitate decomposition of useful metabolites in the absence of a carefully tailored series of linked biochemical steps (see figure 3). Generally, several biochemically coupled reactions with multiple enzymes need to be carefully engineered to work together, with regulatory feedback inhibition, for metabolic processing to work. In this paper we are only considering the difficulty of obtaining a single protein such as an enzyme, and not that of obtaining whole, functional new networks or gene systems.
Systematic modification of parts of a protein

In this approach, a method is needed to efficiently kill off individuals whose mutant protein is not functional. The sequences (usually the base pairs of the gene) present in the survivors are then determined. There are various experimental setups.
In one approach, the original gene is deactivated and the researcher seeks to generate an alternative functional sequence. The protein coded for has a key function, such as being part of a metabolic process to synthesize a necessary biochemical. The researcher keeps the test organism alive by providing the lacking nutrient. Different variants of the defective gene are made available, via a plasmid or other vector, and the nutrient is then denied. Survival indicates a working variant is present.
In another strategy, mutated genes able to protect against a poison or virus are introduced into a host and the sequences from the survivors are analyzed.
Comparison of sequences across taxa
Gene sequences for similar functions across different organisms can be compared in an effort to estimate how much variety is tolerated. Patterns can often be identified, such as that only amino acids possessing similar polarity or size are allowed at a given position on the chain. If the data set is large enough, some rough statement of number of alternatives should be possible.15
Arriving at reliable estimates for a given protein is very difficult. An average protein consists of over 300 AAs, each of which could be affected by mutations with any of 20 possible AAs at each location. Furthermore, one would have to check which mutations are compatible with other mutations on the same gene. Therefore, it is worthwhile to examine carefully the assumptions which the authors use in the estimates reported. This is the purpose of the Appendices. Readers interested in the details are encouraged to read these and to examine the original papers.
The studies discussed in the Appendices explain the basis for the experiments performed to estimate what proportion of amino acid sequences of a particular length would led to the protein function studied. The published numerical estimates are summarized in Table 1, which is the take-home message of this paper.
The astronomically small values are not the probabilities of generating a near-optimal protein or gene, but the chances of generating a starting point before the natural could be invoked. In one paper Dr Heisig and I,16 and in another Drs Scherer and Loewe,17 independently estimated the maximum number of polypeptide alternatives which may have been generated using the most optimistic assumptions possible. The current evolutionary models assume life has existed for about four thousand million years, leading to a large number of organisms which may have generated new genes. Very short generation times, high mutational rates and huge populations were assumed16,17 to provide the largest number of mutational attempts possible to favour the evolutionary scenarios. We estimated that the maximum number of polypeptide variants coded for genetically which could ever have been generated is about 1046.
1046 is the maximum number of attempts available from which the evolutionist must account for all useful proteins produced. Everyone agrees that the vast majority of random polypeptide sequences would be biologically worthless, but the open question is roughly what fraction might be useful.
Systematic studies of gamma-Proteobacteria18,19 show that of about 14,158 gene families present, more than half (7,655) are represented by only one gene. In other words, most of the genes are very different from each other. The common ancestor believed to have lived over 500 million years ago18 did not provide an ancestral evolutionary starting point for all these gene families according to these authors.
Thousands of genes unrelated to both the gamma-Proteobacteria examined and to all others whose sequences are found in public databases must be accounted for. Perhaps they evolved in other organisms and were transferred laterally. However, it was reported19 that 42.5% of the 7,655 single-gene families were unrelated to any other sequences in all current databases.
The general pattern I observe in the literature is that as the number of organisms sequenced increases, ever more unique genes are discovered which are unrelated to any other known genes. Stover et al. were unable to find homologs for 32% of the Open Reading Frames (probable genes) identified in the bacterium Pseudomonas aeruginosa.18,20
The general view among evolutionists is that gene duplication has led to new genes among eukaryotes, but lateral gene transfer (LGT) does this for prokaryotes. The latter represent the vast majority of organisms. But invoking LGT does not solve the problem. Thousands of novel genes, unrelated to others, must come from somewhere.
This analysis shows that none of the examples summarized in Table 1 can be expected to have arisen by chance processes and then fixed in a population. 1046 random attempts are far too few to satisfy the miniscule probabilities calculated. Furthermore, LGT from unknown taxa is often invoked as the origin of novel genes. This implies that the value of 1046 is far too great, since we must subtract the statistical contribution during hundreds of millions of years by the populations assumed to being the LGT recipient. And there are hundreds of additional proteins in all free-living organisms even less likely to have arisen by chance than indicated by most of the probabilities reported in Table 1. I intend to quantify more examples in future papers. That all of these actually arose naturalistically is not reasonable.
Systematic genomic comparison studies are leading to the view18,19 among evolutionists that a core of about 100 unrelated genes are present in all organisms. These alone were insufficient to support a free-living cell, but after countless mutations or gene eliminations all evidence for them has been lost. In any event, it is simply inescapable that at some point a large number of unrelated genes need to have arisen from among random sequences.
I hope Table 1 will provide a good basis for quantitative discussions as to whether design or natural processes best explain the origin of life and the complexity observed. Evolutionary theory assumes that a series of genes evolved from preceding ones. Where the original ones came from fades into the misty zones of speculation. This line of reasoning only makes sense if chains of successive genes, with novel functions, can be built using statistically plausible jumps. The analysis of sequence variability reported here suggests that huge statistical gaps often separate islands of functional proteins from potential starting points.
Appendix A
AroQ chorismate mutase21


The probability of obtaining a functional Chorismate mutase from among 95 amino acid chains was reported in Table 1 as being 10–44. The details of this experiment are summarized in this appendix.
In the experiment21 plasmids containing variants of AroQ chorismate mutase (figures 4 and 5) were introduced into an Escherichia coli strain (KA13). The purpose of the encoded protein is to catalyze the Claisen rearrangement of chorismate to phephenate (figure 6),22 which is an essential step in the biosynthesis of the amino acids tyrosine23 and phenylalanine.24
The DNA sequence was modified in two regions which code for α-helices, engineered in such a manner that only any of eight natural amino acids could appear in those regions. Specifically, every polar amino acid in the original wild type from Methanococcus jannaschii was randomly replaced by one of the four polar natural amino acids, and each non-polar position by one of the four non-polar amino acids. Several positions known to be critical for the enzymatic function were left unchanged.
The modified bacteria were transferred to a minimal medium lacking tyrosine and phenylalanine. The AroQ DNA sequences of the surviving colonies were analyzed and the number of unique variants determined. The authors then extrapolated to conclude that the chances of obtaining a minimally functionally AroQ would be about 1 out of 5 x 1023 sequences from among those generated. In the generated set, however, portions of the three α-helices were replaced by only 8 selected amino acids25 although any of 20 natural amino acids could show up in nature.
Professor Scherer pointed out26 that in these sequences the hydropathy requirements to produce the folds had already been designed into the experiments. This improved the chances of obtaining a functional mutant significantly. To take this into account, he proposed an additional average probability of 0.5 per residue position for the 77 amino acids involved in the α-helices. This leads to a more accurate probability of 0.577 x (2 x 10–24) = 10–47 of obtaining AroQ functionality from among all random polypeptides of the same length as the wild type. Another author suggested a value of 10–53.27,28
Dr Axe has pointed out21 that most proteins are near an optimal state and this needs to be taken into account in these kinds of experiments. Typically certain amino acids must be present and in a very demanding 3-dimensional structure to create an enzymatic active site. Replacing one of these residues can be deadly. The rest of the protein must provide a stable scaffold, which holds the critical portions of some amino acids in ideal locations in three dimensions, for the enzyme to work. Modification in the position of some bonds by a few tenths of an Angstrom is often unacceptable in some regions of a protein.

However, enzymes typically fold reliably into one of the most thermodynamically stable configurations, and this final state is so stable that alternate amino acids often have little effect. One could replace a small number of amino acids at different positions in a large number of separate experiments and then incorrectly conclude that all these substitutions are always permissible in the presence of each other. This is the error of overlooking ‘context dependence’,29–31 also discussed32 in this journal. Taking these factors into account indicates that the estimated proportion of 10–47 is probably too large.
Appendix B
TEM-1 penicillinase27
The probability of obtaining a functional TEM-1 penicillinase from among 153 amino acid chains was reported in Table 1 as being 10–77. The details of this experiment are summarized in this appendix.
β-lactamases are enzymes which protect bacteria from penicillin-like antibiotics. TEM-1 penicillinase is a typical class A β-lactamase consisting of 263 residues, and includes two structural domains. The whole protein, once folded, reveals several features which include nine strands, twelve helices and three chains. The larger 153 amino acid domain was studied by Axe.27
How many sequences would provide the enzymatic function? All possible mutations would require 20153 different genes to be examined, which is not realistic. Axe shows how careful reasoning does permit extrapolation to a reasonable estimate based on far fewer mutants.
Protein folding is a concerted effort involving multiple portions of the polypeptide concurrently. Interactions between the side chains of different amino acids bring portions together in an orchestrated order, which leads reliably to the same three-dimensional, final, stable folded pattern. These considerations imply that the number of distinct folding patterns is relatively small33 and in the order of 103 to 104. This places constraints on the properties of amino acids which may be substituted via mutations.
Alignment of 44 large-domain sequences from different organisms, obtained from public databases, allowed each of the 153 positions to be characterized in terms of the properties of the amino acids tolerated there: hydrophobic, hydrophilic, intermediate, not hydrophobic, not hydrophilic or unconstrained. This defined the hydropathic signature of this protein folding class (figure 7).
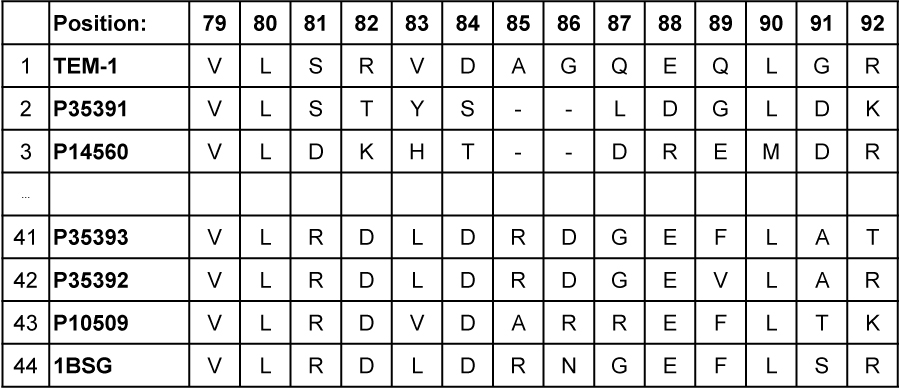
Proteins often continue to function in spite of mutations due to excess robustness built in. Portions of the folded chain are held together near optimally, under thermodynamical considerations, through a large number of interactions. Therefore, sub-optimality through a few mutations will often not lead to discernable loss in function. Therefore, one cannot conclude that mutations which individually seem harmless would be acceptable when present concurrently. The optimized proteins have a kind of ‘buffering’ effect. Demonstrating that alternative amino acids are acceptable, by inducing mutations on a near optimal wild type, does not permit an estimate of the number of acceptable sequences with minimal functionality. To make a reasonable estimate would require actually generating the variants with multiple mutations to identify which alternatives would really work.
The design of Axe’s experiment27 reflects how natural selection would have to go about fine-tuning a novel enzyme. A minimally useful sequence must first exist upon which natural selection could act. He generated a large number of TEM-1 variants by mutating 49 positions, introduced the plasmids in an E. coli strain by electroporation, and isolated a colony having 33 substitutions (relative to the original sequence). Exposure to a low concentration of ampicillin permits selection of those bacteria with a functioning enzyme. The candidate starting sequence for the subsequent experiments showed resistance at 10 μg/ml, but none at 20 μg/ml concentrations at 25°C.34 This enzyme provided 0.3% of the wild type activity at 25°C, and only 0.01% at 37°C.34 Since the enzymatic reactive site was not mutated, the loss in activity probably reflects lower ability to hold the protein together in a suitable three-dimensional geometry.
A large number of bacteria with the above less-than-optimal candidate starting sequences were grown. From these sequences, mutant plasmids where engineered in a manner to optimize the proportion satisfying the hydropathic signature. Four sets of random mutations, each involving ten residue positions, were performed. The number of mutants satisfying the hydropathic signature was calculated (on average over 85% of the mutants generated), and those surviving ampicillin poisoning were sequenced. The geometric mean calculated from the pass rates of the four experiments led to an upper-bound estimate of 0.38 per position. This is the probability that a random mutation at a residue position which meets the hydropathic signature constraints will be acceptable.
The value of 0.38 is generous for several reasons. In one of the four experiments no acceptable mutants were obtained (of 54,000 mutants generated which still satisfied the hydropathic signature!), but a probability of 0.002 was used anyway. Furthermore, acceptable mutations within sets of ten residue positions will certainly not be permissible in the presence of all other acceptable mutations for the remaining 153–10 positions.

For the whole large domain (figure 8) the proportion of acceptable mutants which are signature compliant would thus be less than 0.38153 = 10–64.35 The number of open reading frames (here only a portion of a gene) leading to the signature under study, based on which codons code for which amino acids, is 10–33. In conclusion, among random polypeptides a proportion of less than 10–64 x 10–33 = 10–97 would provide a working large domain β-lactamase enzyme using the same fold characteristics.35
It is possible other protein folding families could offer a necessary stable framework for enzymatic activity. If a species has about a million different protein variants and a thousand or so fold types, then about 0.1% of the fold types on average would be suitable for a particular function. Based on other work,36 at most 1 out of 1010 random sequences would fold to a stable pattern based on hydropathic constraints alone. These considerations led to an estimate28 that about 1 out of 1077 sequences of 153 amino acids could perform the function under study.
Appendix C
Sequence analysis of the lambda repressor fold36The probability of obtaining a functional lambda repressor from among 92 amino acid chains was reported in Table 1 as being 10–63. The details of this experiment are summarized in this appendix.
Background
Bacteriophage lambda, probably the most extensively studied bacterial virus,37 has a genome of about 50 genes,38 and under suitable conditions can become integrated into DNA of bacteria such as E. coli. Within the host there are two modes of replication.39 (1) Once integrated into the host genome it can be replicated along with the rest of the DNA. A key component of this prophage state is the lambda repressor protein (cI protein), which occupies the operator, blocking the alternative reproductive pathway, and also activates its own transcription. (2) In the lytic state, whereby the virus is not inserted into the host chromosome, the cro protein attaches to a different site in the operator, preventing synthesis of the repressor protein and permitting its own synthesis.
In the prophage state most of the virus genes are not transcribed. In the lytic state the virus DNA is extensively transcribed and organized into new bacteriophage, then released by rupturing the host cell’s outer membrane. This kills the cell, of course.
An infecting virus usually adopts the prophage stage. But when the host is badly stressed or damaged, an integrated virus converts to the lytic state. For this to be possible, the repressor protein needs to be inactivated.
The lambda repressor protein is an example of helix-turn-helix proteins which bind to specific DNA sequences.40 Other examples include tryptophan repressor, lambda cro and CAP.41 These kinds of proteins often exist as symmetric dimers, able to bind to two DNA stretches per protein (e.g. on opposite strands of complementary DNA), which doubles the number of contacts and squares the affinity constant.41
A mutant variant of lambda repressor protein not able to function properly is easy to monitor experimentally, since the virus in the lytic state kills the host.
The experiments
Sauer at MIT examined42 mutants at 25 residue positions (8–23 and 75–83) in two α-helical regions of the γ-repressor distributed along positions 1 to 92 of the N-terminal end of the protein. The whole protein usually contains about 237 residues.43–45 Plasmids were engineered which contained an ampicillin-resistant gene and an origin of replication which allows production of single-stranded DNA for sequencing purposes.
Oligonucleotide cassettes46 were synthesized for several experiments. At each position where amino acid mutations in the protein are to appear, codons of type NNG/C were prepared in equal proportions, where the N indicates any of the four bases (A, C, T or G). Thus only 32 of the possible 64 genetic code alternatives were needed to generate all possible natural amino acids. Between one and three positions were allowed to differ from the wild type.
The modified plasmids contained special restriction sites which permitted the cassettes to be ligated at predetermined positions, ensuring the desired mutant proteins would be coded for. The plasmids were transformed into E. coli K-12 strain X90. Exposure to ampicillin killed off the E. coli lacking inserted plasmid (since the bacteria lacks the ampicillin-resistant gene provided via the plasmids). The phages’ cI then destroy the cells lacking a suitable γ-repressor, since the virus only had the option of entering the lytic state. Surviving E. coli colonies thus have functional repressor protein present in the plasmid. At least 5–10% of wild-type activity was necessary to survive.

Survivors were analyzed and the alternative amino acids at each residue position were reported. The 25 positions mutated were supplemented with the results from an earlier study42 in which positions 84–91 had been mutated in three separate experiments involving three and four residue positions at a time. The alternative amino acids found at each residue position are shown in figure 9.
The available data gives some indication as to the tolerable variability. By multiplying the number of alternatives at each position shown in figure 9 the authors concluded that about 4 x 1022 different sequences would be functional over the 33 positions.47 Extrapolating to the 92 positions of the domain under consideration indicates that a proportion of about 10–63 would be functional.
I believe this estimate is still too large due to context dependence: a tolerated mutation at one position will often be deactivating when multiple other otherwise acceptable mutations are present. At one point they write,
‘However, in general there appears to be no dependence of a change at one position on a change at another, as most changes were recovered in several different mutant backgrounds.’48
This is a surprising statement for several reasons. What is needed are experiments in which only mutations at a particular residue are generated, followed by additional tests for which this and additional residues are modified.


In the reported data43 only two such series of experiments were performed, generating at most three mutations with respect to the wild type. This permits us to determine whether the same mutations at one position affect the probability of additional ones being acceptable elsewhere.
(i) All possible mutations were generated in positions 14, 15/16 and 14–17. The results are shown in figure 10. Unfortunately, no variability was found in position 14, so this is uninformative. Experiment ‘14–17’ produced the wild type sequence and one mutation (R→K) at position 17. Experiment ‘15/16’ also produced the wild type sequence and four other amino acids were tolerated at position 16. But why were none of these four alternatives at that position identified in experiment ‘14–17’? The extra mutation, (R→K), probably hindered this!
(ii) All possible mutations were generated in positions 83, 81/82 and 81–83.
The results are shown in figure 11. Experiment ‘83’ generated the wild
type plus ten other amino acids at that position. Experiment ‘81–83’,
however, failed to produce six of these at the same position 83. This may well be
due to the presence of additional mutations at the other two locations. It is also
true, that experiment ‘81–83’ did not produce any functional variants
with an amino acid missing at the corresponding site of experiment ‘83’.
In most cases an A is found at residue 81 for experiments ‘81/82’ and
‘81–83. Lets examine these cases. Experiment ‘81/82’ displayed,
in addition to the wild type R at position 82, another 11 alternative amino
acids. Of these, experiment 83 produced only 1 (E)! It would appear, once
again, that mutations in the third position limited the number of possibilities
at other residues.33
(iii) Experiments from the same laboratory were reported earlier in which seven positions of the same protein were mutated. I pointed out in this journal32 that the number of variants generated increased greatly with number of mutational differences from the wild type. Nevertheless, the number of functional alternatives identified was precisely in the opposite order. Clearly, on average, existing mutations prevented otherwise acceptable mutations from producing a functional protein.
This fact is surely known to these researchers, since in the earlier paper the authors wrote:
‘In separate experiments, five of the seven core positions were altered individually. Only one to three amino-acid substitutions at each position yield a fully functional protein … as is common for buried positions.’49
The authors of the later paper36 are aware that the context of mutations are an issue, and pointed out with respect to the estimated number of acceptable sequences,
‘On the one hand, this calculation overestimates the number of functional sequences, since changes at individual positions are less likely to be independent of one another as more positions are allowed to vary. Moreover, combining changes at several positions, each of which individually decreases the activity of the proteins slightly, may result in a protein that is essentially non-functional. On the other hand, some changes which are not allowed when positions are randomized individually may be tolerated in other sequence contexts.’50
However, the magnitude of both effects is surely dramatically different and hardly compensate significantly. What examples for novel compensatory multiple mutations are found in the data reported?42 At most only one. In experiment ‘81–83’ amino acids SA in the first two positions led to a functional protein, but this mutant was not found in experiment ‘81/82’ (an example was obtained with SR). On the other hand, the authors pointed out51 that there is a 58% chance that not all tolerated amino acids were identified at position 82, making likely that a larger data set for experiment ‘81/82’ may well display the ‘missing’ amino acid.
Whether introducing simultaneously multiple mutations which compensate for each other is actually realistic to evolutionary theory, is questionable. For example, it is possible that using the two largest hydrophobic and a single smallest hydrophobic residue would work in some context, but whether theoretical intermediates (e.g. one of the largest hydrophobic amino acids only) might work is not assured. Such solutions would often require an all-or-nothing set of circumstances.
In contrast, an overly generous assumption of mutational context independence can have a dramatic effect. Let us reconsider the data in figure 11 and neglect the few sequences for which an A was not found in position 81. Experiment ‘83’ produced ten alternatives, and experiment ‘81/82’ generated eleven functional alternatives at position 82. We see that this simplification reflects closely the assumptions made in figure 9 regarding residues 82 and 83. Then the assumption of context independence, as proposed by the authors, predicts about 10 x 11 = 110 variants from experiment ‘81–83’ (with a wild type A in position 81), or 1101/2 = 10.49 per position properly weighted. However, only 4 were actually found, 41/2 = 2 on average. Whether one assumes (10.49)n or (2)n over n residue positions, leads to dramatic different estimates for the number of acceptable variants.
Testing all mutations at a large number of positions is experimentally not feasible, given the huge number of possibilities 20n for n residues positions. Simplifying approaches are needed leading to large doubts in the estimates. The proposal of about 1057 functional alternatives50 seems to be too high, since for this to be possible up to 67 of the 92 positions of this portion of the protein must be mutable at the same time and in all combinations based on the data from figure 9. (In the 33 residues studied 9 positions were invariant (see figure 9), so all but 9 x 92/33 positions on the full domain must tolerate for all the combinations of mutations to arrive at the authors’ number of polypeptide alternatives, 1063).
A more realistic estimate might be ‘guesstimated’ as follows. We shall assume that any of the mutations shown in figure 9 are acceptable and also concurrent. The authors used a Monte Carlo simulation to identify the probability that not enough plasmids had been generated to identify all acceptable alternatives at each position. For all those residues51 we shall assume two more amino acids would be acceptable (Table 2), and shall further assume that all these additional mutations would be mutually compatible. We neglect the possibility of a handful of variants involving multiple compensatory mutations. As shown in Table 2, about 3.1 x 1021 alternatives were estimated.

The number of alternatives will be extrapolated by a simple factor of 92/33 to cover the whole domain, i.e. about three separate sections. As a partial compensation for the above assumptions, we will say that acceptable mutations are limited to each of the roughly three sections, but not between them. This leads to an estimate of (92/33) x (3.1 × 1021) = 8.6 × 1021.
The resulting proportion of functional variants, 8.6 x 1021 / (20)92 (ca. 2 × 10–98) is considerably smaller than what the authors suggested, 10–63.
Appendix D
Cytochrome c proteins52
The probability of obtaining a functional cytochrome c from among amino acid chains of suitable length was reported in Table 1 as being 10–44 in one case and 10–112 on another. The details of this experiment are summarized in this appendix.
Yockey52 collected a list of all known cytochrome c protein sequences and lined up 110 residue positions after taking into account apparent mutational deletions. He then expanded the list of known sequences generously, using a model53 developed by Borstnik and Hofacker,54,55 assuming many other sequences might also be tolerated, as already discussed in this journal.16 We reported56 that a fraction in the order of 2.0 x 10–44 of the 110-residue chains would offer a starting point for natural selection to begin fine-tuning a cytochrome c sequence. Incidentally, the information theory basis for these calculations assumes context independence:32 all individually acceptable amino acids substitutions would supposedly lead to a functional cytochrome c as in the presence of other mutations. The true proportion of functional alternatives is surely many orders of magnitude smaller, a mathematical issue in the use of information theory I have brought to Yockey’s attention.
Yockey’s latest calculations57 suggested that the proportion of polypeptides leading to functional cytochrome c is actually much lower: 1.6 x 10–112.
Appendix E
Ubiquitin58This protein is present in all examined eukaryotes type cells. Current evolutionary thinking is that the first eukaryote cell lived about 2.7 Ga ago.48 Since all plants, animals and fungi possess ubiquitin (UB), unlike prokaryotes, this gene must have arisen virtually instantaneously under evolutionary assumptions.58
I collected all known and reliable sequences for UB and calculated the number of alternatives using information theory. About 60% of UB residues seem to tolerate no mutations at all, and in 17 other positions a single alternate amino acid was occasionally found. In almost all the latter cases this exception was found in only a single organism, and some of these sequences may simply be incorrectly reported data.
I estimated58 that a proportion of about 4 x 10–83 polypeptides, 76 residues long, would produce a functional UB. Several things need to be considered in this estimate.
(i) Not all eukaryotes have been examined. On the other hand, many sequences were identical for species not especially close according to current evolutionary theory. Therefore, not too much more variety is to be expected.
(ii) There are at least three families of ubiquitin distinct for animals, plants and fungi. It is possible these alternatives are not interchangeable, in which case the amount of acceptable variability for the putative initial ancestor would be restricted.
(iii) My estimate assumes that all mutations present at any residue would be compatible with all other mutations at other locations.
References
- In addition to the 20 commonly used natural amino acids, at least two more are known to be coded for genetically in small amounts in a few organisms. Return to text.
- For example, many French scientists, sometimes due to a philosophical background in vitalism, are very hostile to the possibility that a personal God exists. Remy Chauvin ravaged Darwinian theory in Le darwinisme ou la fin d’un mythe, Editions du Rocher, 1997. I know personally several prominent members in the Intelligent Design movement who do not subscribe to belief in any kind of deity. Their conviction that neo-Darwinian processes are unworkable and dismay at the rampant dogmatism in which the opposite is claimed, has led them to join the movement to force an open discussion in the academic world. Return to text.
- Richard Dawkins argues in The Blind Watchmaker, Penguin Books, London, 1986, and elsewhere that a simple ‘replicator’ can self-refine through Darwinian processes over time. Return to text.
- The phrase ‘RNA world’ is generally attributed to Harvard University’s Walter Gilbert: The RNA world, Nature 319:618, 1986. Return to text.
- Eigen, M. and Schuster, P., The Hypercycle: A Principle of Natural Self-Organization, Springer Verlag: Berlin, 1979. Return to text.
- Ohno, S., Evolution by Gene Duplication, Springer Verlag, New York, 1970. Return to text.
- Davidson, A.R. and Sauer, R.T., Folded proteins occur frequently in libraries of random amino acid sequences, Proc. Natl. Acad. Sci USA 91:2146–2150, 1994. Return to text.
- Davidson, A.R., Lumb, K.J. and Sauer, R.T., Cooperatively folded proteins in random sequence libraries, Nature Struct. Biol. 2:856–863, 1995. Return to text.
- Keefe, AD and Szostak, J.W., Functional proteins from a random-sequence library, Nature 410:713–718, 2001. Return to text.
- Yamouchi, A., Nakashima, T., Tokuriki, N., Hosokawa, M., Nogamai, H., Arioka, S. et al., Evolvability of random polypeptides through functional selection within a small library, Protein Eng. 15:619–626, 2002. Return to text.
- Hayashi, Y., Sakata, H., Makino, Y., Urabe, I. and Yomo, T., Can an arbitrary sequence evolve towards acquiring a biological function? J. Mol. Evol. 56:162–168, 2003. Return to text.
- Tsuji, T., Kobayashi, K. and Yanagawa, H., Permutation of modules or secondary structure units creates proteins with basal enzymatic properties, FEBS Letters 453:145–150, 1999. Return to text.
- Wagner, A., Energy Constraints on the Evolution of Gene Expression, Mol. Biol. Evol. 22(6):1365–1374, 2005. Return to text.
- Glickman, M.H. and Ciechanover, A., The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction, Physiol Rev. 82:373–428, 2002. Return to text.
- This assumes all protein variants are functional in all organisms involved in the comparison. Return to text.
- Truman, R. and Heisig, M., Protein families: chance or design? Journal of Creation 15(3):115–127, 2001. Return to text.
- Scherer, S. and Loewe, L., Evolution als Schöpfung? in: Weingartner, P. (Ed.), Ein Streitgespräch zwischen Philosophen, Theologen und Naturwissenschaftlern, Verlag W. Kohlhammer, Stuttgart; Berlin; Köln: Köhlhammer, pp. 160–186, 2001. Return to text.
- Lerat, E., Daubin, V. and Moran N.A., From gene trees to organismal phylogeny in prokaryotes: the case of the g-Proteobacteria, PloS Biology 1(1):101–109, 2004. Return to text.
- Lerat, E., Daubin, V., Ochman, H. and Moran, N.A., Evolutionary origins of genomic repertoires in bacteria, PolS Biology 3(5):0807–0814, 2005. Return to text.
- Stover, C.K., Pham X-QT., Erwin, A.L., Mizoguichi, S.D., Warrener, P. et al., Complete genome sequence of Pseudomonas aeruginosa PA01, an opportunistic pathogen, Nature 406:959–964, 2000. Return to text.
- Taylor, S.V., Walter, K.U., Kast, P. and Hilvert, D., Searching sequence space for protein catalysts, Proc. Natl. Acad. Aci. Sci. USA 98(19):10596–10601, 2001. Return to text.
- Stryer, L., Biochemistry, W. Freeman and Company, New York, Fourth Ed., p. 725, 1999. Return to text.
- <http://pathway.yeastgenome.org:8555/YEAST/new-image?type=PATHWAY&object=TYRSYN&detail-level=3>, 30 August 2005. Return to text.
- <http://pathway.yeastgenome.org:8555/YEAST/new-image?type=PATHWAY&object=PHESYN&detail-level=3>, 30 August 2005. Return to text.
- One of the α-helices was modified in one experiment and the next two α-helices were both modified in a second experiment. Survivors from both experiments were combined in a third experiment. In the latter case, the new genes possessed only mutated versions of the α-helices which individually were functional. Return to text.
- Scherer, S., In search for the prevalence of enzymatically active structures in amino acid sequences space, Tagungsband der 22. Fachtagung für Biologie 11:41, 13 March, 2005. Return to text.
- Axe, D.D., Estimating the prevalence of protein sequences adopting functional enzyme folds, J. Mol. Biol. 341:1295–1315, 2004. Return to text.
- Axe, ref. 27, p. 1310. Return to text.
- Axe, D.D., Foster, N.W. and Fersht, A.R., Active barnase variants with completely random hydrophobic cores, Proc. Natl. Acad. Sci. USA 93:5590–5594, 1996. Return to text.
- Axe, D.D., Extreme functional sensitivity to conservative amino acid changes on enzyme exteriors, J. Mol. Biol. 301:585–596, 2000. Return to text.
- Axe, D.D., Foster, N.W. and Fersht, A.R., A search for single substitutions that eliminate enzymatic function in a bacterial ribonuclease, Biochemistry 37:7157–7166, 1998. Return to text.
- Truman, R., Protein mutational context dependence: a challenge to neo-Darwinian theory: part 1, Journal of Creation 17(1):117–127, 2003. Return to text.
- Lim, W.A. and Sauer, R.T., Alternative packing arangements in the hydrophobic core of l repressor, Nature 339:31–36, 1989. Return to text.
- Axe, ref. 27, p. 1300. Return to text.
- Axe, ref. 27, p. 1308. Return to text.
- Lau, K.F. and Dill, K.A., Theory for protein mutability and biogenesis, Proc. Natl. Acad. Sci. USA 87:638–642, 1990. Return to text.
- Lodish, H., et al., Molecular Cell Biology, Second Printing, W.H. Freeman and Company, New York, pp. 216–219, 2000. Return to text.
- Alberts, B., Johnson, A., Lewis, J., Raff, M., Roberts, K. and Walter P., Molecular Biology of the Cell, Third Ed., Garland Publishing, New York, 1994. Return to text.
- Alberts et al., ref. 38, p. 443. Return to text.
- Harrison, S.C. and Aggarwal, A.K., DNA recognition by proteins with the helix-turn-helix motif, Annu. Rev. Biochem. 59:933–969, 1990. Return to text.
- Alberts et al., ref. 38, p. 409. Return to text.
- Reidhaar-Olson J.F. and Sauer R.T., Functionally acceptable substitutions in two α-helical regions of γ-repressor, Proteins: Structure, Function, and Genetics 7:306–316, 1990. Return to text.
- Perna, N.T. et al., Genome sequence of enterohaemorrhagic Escherichia coli O157:H7, Nature 409(6819):529–533, 2001. See also: <www.ncbi.nlm.nih.gov/BLAST/Blast.cgi> Accession AAG54571 Version AAG54571.1 GI:12513020 for the sequence. Return to text.
- Juhala, R.J., Ford, M.E., Duda, R.L., Youlton, A., Hatfull, G.F. and Hendrix, R.W., Genomic sequences of bacteriophages HK97 and HK022: pervasive genetic mosaicism in the lambdoid bacteriophages, J. Mol. Biol. 299(1):27–51, 2000. See also <www.ncbi.nlm.nih.gov/BLAST/Blast.cgi> Accession AAF31095 Version AAF31095.1 GI:6901592 for the sequence. Return to text.
- Clark, A.J., Inwood, W., Cloutier, T. and Dhillon, T.S., Nucleotide sequence of coliphage HK620 and the evolution of lambdoid phages, J. Mol. Biol. 311(4):657–679, 2001. See also <www.ncbi.nlm.nih.gov/BLAST/Blast.cgi> Accession AAK28868 Version AAK28868.1 GI:13517579 for the sequence. Return to text.
- Reidhaar-Olson, J.F., Sauer, R.T., Combinatorial cassette mutagenesis as a probe of the informational content of protein sequences, Science 241:53–57, 1988. Return to text.
- Reidhaar-Olson and Sauer, ref. 42, p. 315. The statement ‘for the 30 residue positions’ seems to be a typographical error, 33 was meant. Return to text.
- Hedges, S.B., Blair, J.E., Venturi, M.L. and Shoe, J.L, A molecular timescale of eukaryote evolution and the rise of complex multicellular life, BMC Evolutionary Biology 4:1–9 (2004). See also <www.biomedcentral.com/1471-2148/4/2#B1>, 23 December 2005. Return to text.
- Lim and Sauer, ref. 33, p. 32. Return to text.
- Reidhaar-Olson and Sauer, ref. 42, p. 315. Return to text.
- Reidhaar-Olson and Sauer, ref. 42, p. 313. Return to text.
- Yockey, H.P., Information Theory and Molecular Biology, Cambridge University Press, Cambridge, 1992, p. 250. Return to text.
- Yockey, ref. 52, p. 136. Return to text.
- Borstnik, B. and Hofacker, G.L.; in: Clementi, E., Corongiu, G., Sarma M.H. and Sarma, R.H. (Eds.), Structure & Motion, Nucleic Acids & Proteins, Guilderland, Adenine Press, New York, 1985. Return to text.
- Borstnik, B., Pumpernik, D. and Hofacker, G.L., Point mutations as an optimal search process in biological evolution, J. Theoretical Biology 125:249–268, 1987. Return to text.
- Truman and Heisig, ref. 16, p 117. Return to text.
- Yockey, H.P., Information Theory, Evolution, and The Origin of Life, Cambridge University Press, Cambridge, chapter 6, 2004. Return to text.
- Truman, R., The ubiquitin protein: chance or design? Journal of Creation 19(3):116–127, 2005. Return to text.

Readers’ comments
Comments are automatically closed 14 days after publication.