

Journal of Creation 21(2):90–100, August 2007
Browse our latest digital issue Subscribe
Genetic code optimisation: Part 1
The genetic code as we find it in nature—the canonical code—has been shown to be highly optimal according to various criteria. It is commonly believed the genetic code was optimised during the course of an evolutionary process (for various purposes). We evaluate this claim and find it wanting. We identify difficulties related to the three families of explanations found in the literature as to how the current 64 → 21 convention may have arisen through natural processes.

The order of amino acids in proteins is determined by information coded on genes. There are over 1.51 × 1084 possible1 genetic codes based on mapping 64 codons to 20 amino acids and a ‘stop’ signal2 (i.e. 64 → 21). The origin of code-based genetics is for evolutionists an utter mystery,3 since this requires a large number of irreducibly complex machines: ribosomes, RNA and DNA polymerases, aminoacyl tRNA synthetases (aaRS), release factors, etc. These machines consist for the most part of proteins, which poses a paradox: dozens of unrelated proteins are needed (plus several special RNA polymers) to process the encoded information. Without them the genetic code won’t work, but generating such proteins requires that the code already be functional.
This is one of many examples of ‘chicken-and-egg’ dilemmas faced by materialists. Another is the need for a reliable source of ATP for amino acids to polymerise to proteins: without the necessary proteins and genes already in place such ATP molecules won’t be produced. In addition, any genetic replicator needs a reliable ‘feed stock’ of nucleotides and amino acids, but several of the metabolic processes used by cells are interlinked. For example, until various amino acid biosynthetic networks are functional, the nucleotides can’t be metabolised. These are some of the reasons we believe natural processes did not produce the genetic code step-wise. We hope to present a detailed analysis of the minimal components needed for a genetic code to work in a future paper, but this is not the topic we wish to address here.
The literature is full of papers which claim the universal code4 has evolved over time and is in some sense now far better than earlier, perhaps even near optimal. We cannot address all the models and claims here, but we hope to present a few thoughts which we hope will show that these claims are ‘flights of fantasy’. No real workable mechanism has yet been offered1,3 as to how a simpler genetic system could have increased dramatically in complexity and in robustness towards mutations. If a primitive replicator had gotten started, contra all chemical logic, would it be possible according to various evolutionary scenarios to refine the system to generate the 64 codon → 20 amino acid + ‘stop’ signal convention used by the standard genetic code?
Origin of any genetic code
Before an evolutionary process could optimise a code, a replicating lifeform must first exist with some kind of information processing capabilities. Trevors and Abel published one of the most honest and illuminating papers3 on the issues which confront a naturalistic explanation for the origin of life. In particular the origin of an information storing and processing system, able to guide the synthesis of proteins, is recognized as incomprehensible. In their own words, ‘Thus far, no paper has provided a plausible mechanism for natural-process algorithm-writing’.5 Abel is well known for his attempts to find a natural origin for the genetic code and naturalistic explanation of the origin of life. He and The Origin-of-Life Foundation, Inc. ® have a standing offer of $1 million to anyone providing a plausible natural solution.6 In stark contrast to the straightforward honesty offer in this paper3 are a large number of Origin-of-Life papers which appeal to no recognizable chemistry and offer no conceptually feasible path as how to go from their vague notions to extant genetic systems.
There are three basic approaches7 used by materialists to explain the 64 → 21 mapping of the genetic code: (I) chemical/stereochemical theories, (II) coevolution of biosynthetically related amino acid pathways and (III) evolution and optimisation by natural selection to prevent errors. There is a logic to the order in which we present these three approaches. (I) is closest to the question of a natural origin for a biological replicator. (II) already requires a large number of complex and integrated biochemical networks to be in place. Attempts to explain the 64 → 21 code mapping at this level would clearly mean ignoring the question as to where all these molecular machines and genes came from. (III) Evolutionary hypotheses to explain the 64 → 21 mapping at this level would require assuming all 20 amino acids are already present in a genetic code and that most genes already code for highly optimised proteins.
(I) Chemical/stereochemical theories
All the suggestions in this area assume some kind of simple starting system, being guided by natural chemical processes. These primitive systems then accumulated vast amounts of complexity and sophistication.
Attempts have been made to find direct chemical interactions between portions of RNA and amino acids.8 These are supposed to have led to the genetic code. Amino acids might bind preferentially to their cognate codons,9 anticodons,10 reversed codons,11 codon-anticodon double helices12 or other chemical structures.
After admitting that ‘there is little evidence for selective binding of amino acids to isolated codons or anticodons’, Alberti13 proposed that chains of mRNA would interact with special tRNA chains, and short peptides would attach specifically to these tRNAs. Being now brought close together, the short peptides would polymerise to form proteins. A number of cofactors would stabilize the tRNA-mRNA interactions, eventually becoming ribosomes. Another set of cofactors would decrease the number of amino acids needed to provide a specific interaction with the various tRNA, which today is done by aaRSs.
Objections. None of the reports in this area reveal any kind of consistent association between codons and the amino acid expected based on the genetic code.14 The wide variety of chemical systems intelligently conceived in the various scenarios cannot be justified for free nature conditions, and excessive freedom exists in the interpretation of such models, undermining the significance of any particular one.7 Therefore, it is often alleged15 that the original chemical interactions can no longer be identified through the present coding assignments of the genetic code, but that such putative interactions may have gotten the process started.16
Amino acids created under abiotic conditions are assumed to have been introduced first in a primitive code.17 But, since all but glycine come in d and l mirror-image forms18 such a source of amino acids would lead to chaos. In addition, the 3 chiral C atoms in ribose in RNA would produce even more stereoisomers in free nature. Furthermore, claiming17,19 that the amino acids found in the Miller experiment would have been the first to be used by a genetic code makes a dope20 out of the reader who accepts this, since geologists today believe the gases used in such experiments have no relevance to a putative early atmosphere.18,21,22 Subsequent experiments with more reasonable gas mixtures generated very little organic material and virtually no amino acids at all.18,23,24
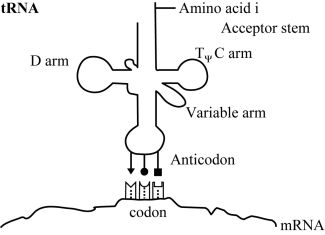
At this time, the order in which amino acids are to polymerise is not communicated by the genetic code through direct amino acid interactions with DNA or RNA polymers. Transfer RNA is used to map codons to their specific amino acids. Three specific nucleotides (the anticodon) are part of the tRNA molecules, and these interact transiently with their cognate codons on mRNA. In figure 1 we show how specific codon-anticodon interactions determine which amino acid is coded for by a mRNA nucleotide triplet. The codon-anticodon interactions must be weak enough to permit separation once no longer needed, but with sufficient specificity to prevent incorrect binding. But in the absence of additional machinery such as ribosomes to help hold everything in place, the interactions between codons and the adaptor’s anticodon would be too weak to be of any value. At a distant and physicochemically unrelated portion of the tRNA adaptor a specific amino acid must therefore be attached (with the consumption of a high energy ATP molecule) (figure 1).
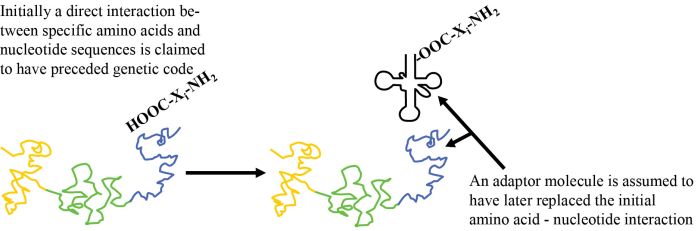
How is nature supposed to have gone from an initial system, involving a chemical or a physical interaction of amino acid i (‘AAi’) (where i represents version 1, 2, 3 …) with RNA tri-nucleotide i (‘codoni’), to the current scheme based on adaptor i (‘adapi’)? Two things must now occur simultaneously (see figure 1). One part of a given adaptor number i, adapi, must replace the original AAi/codoni interaction, and to a second part of adap1 the same AA; must now be attached (figure 2). These cannot occur sequentially, as both kinds of bonds must occur simultaneously if the primitive ‘code’ based on direct interaction is to be retained. Since the spatial relationship with other amino acids is now very different, any putative chemical reactions with other amino acids can no longer occur. This means all the amino acid to template interactions must be replaced simultaneously! One cannot have a mixed strategy, since then only part of the putative original polypeptide could form.
|

|
If the ancestral replicator functioned reliably without an adaptor, the new system using many specialized adaptor molecules must be at least as effective immediately, otherwise the former would out-populate the new evolutionary attempt. This means that attachment of AAi to adapi must be highly reliable, as is the case with modern aminoacyl tRNA synthetases. Among other implications, this requires a reliable source of the different adaptors i=1,2,3 … (adap1) during the ‘lifetime’ of this ‘organism’ and during the subsequent ‘generations’. Specifically, all these adaptor sequences must be immediately metabolized consistently and in large amounts for the new coding scheme to function.
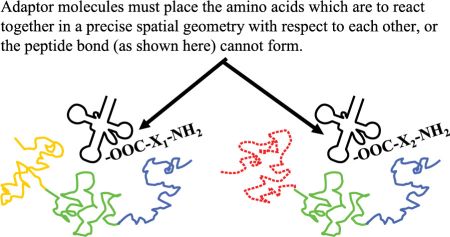
The adaptor molecules must satisfy several structural requirements. The location where amino acid i is attached to its cognate tRNAi must be at an acceptable distance and geometry to facilitate formation of the peptide bond (figure 3). Each kind of adaptor molecule must fold reliably into a consistent three-dimensional structure which is able to bring the reacting amino acids and cognate codons into the correct geometry with respect to each other (figure 4). In tRNAs this is accomplished by strategically located base pairing and RNA strands of just the right length.
Even if two sets of tRNA-amino acid complexes were to be bonded simultaneously somewhere along the template, these won’t form a peptide bond in the absence of the carefully crafted translation machinery. Unless carefully engineered, the adaptors would tangle together with themselves and with the template triplet nucleotides (figure 5). Even if these theoretical adaptors could hold two amino acids close enough to react, the endothermic peptide-forming reaction isn’t going to occur spontaneously. Formation of a peptide bond in living organisms is driven by high-energy ester bonds between amino acids and tRNAs, with the help of aminoacyl tRNA synthetases. Theoretical adaptors which merely hold the reactants physically close together is not sufficient. Should on rare occasions a peptide bond actually form, the resulting molecule would probably remain covalently bonded to one of the adaptors (figure 6) afterwards. One of the design requirements of ribosomes is to move the mRNA along in a ratchet-like manner, detaching the tRNA whose amino acid has already been used. For this purpose energy is provided by GTP, and a complex scheme is used to remove the final polypeptide from the mRNA. This requirement has also been overlooked in the conceptual model presented.

|

|
If, in spite of the above observations, polypeptides were to start forming; intramolecular reactions, in which the carboxyl end portion of one amino acid bonds to the amino group of the other amino acid in a growing chain would dominate (figure 7). This is simply because they are close to each other and would probably react with themselves before other amino acids show up to extend the chain length. The ribosome machinery is designed to prevent this from occurring.

i = 1 to 20, and Xi = CHRi (Ri are the side chains).
Furthermore, peptide bonds involving the side chains of amino acids can also form, leading to complex and biologically worthless mixtures. For example, amino groups (-NHR) are present on the side chains of amino acids tryptophan, lysine, histidine, arginine, asparagine and glutamine and can react with the carboxylic acid (-COOH) groups of other amino acids. This is especially true if hot conditions are assumed25 to permit peptide bonds to form. Conversely, some side chains also have carboxylic acids (aspartate and glutamate), which can form amides with any amino group. The highly complex portions of the ribosome machinery were designed to prevent such undesirable side reactions from occurring, by holding the functional groups precisely in place to guide the peptide reactions, and by isolating the functional groups which are not supposed to react together. This very problem is a real issue with automated peptide synthetic chemistries used today, requiring complex side-chain blocking strategies in order to allow the correct peptide extension reactions.
Alberti, mentioned above,13 introduced a different scenario: the adaptor is part of the genetic apparatus from very early on. Basically, one must assume that mRNAs, ribosomes, amino acids and tRNAs all came together long ago with a minimum of complexity. Then evolution performed a series of unspecified steps approaching the miraculous, resulting in the genetic code. The initial system somehow added a multitude of molecular tools and was relentlessly fine-tuned. Any other evolutionary model based on similar premises would resemble closely in many details what he proposes. The necessary subsequent stages must occur if these assumptions are used. Therefore, it is worthwhile to devote some thought as to whether the various processes could reasonably occur naturally. Our comments necessarily apply to other possible variants of the basic thesis.

The basic notion is shown in figure 8. In practice we will show that virtually none of the necessary claims in such scenarios would work. From start to end, chemical and physical realities are abused.
- Nature does not produce stereochemically pure polypeptide and polyribonucleotide chains. Therefore, there is no way to initiate a minimally functional proto-code. First, there is the problem of the source of optically pure26 starting materials. Second, in an aqueous solution, a maximum of 8–10 RNA-mers can polymerise27 and polypeptide chains would be even shorter, even after optimizing for temperature, pressure, pH, and concentration of amino acid, plus addition of CuCl2 and rapidly trapping the polypeptide in a cooling chamber.28,29 The reactants would be extremely dilute, since the thermodynamic direction would be to hydrolyse back to starting materials.
Alternative, non-aqueous environments, such as the side of a dry volcano, would be chemically unpromising. If optically pure nucleotides and amino acids were present, under dry, hot reaction conditions, then larger molecules would form. But the result would be ‘gunk’ or tar, since a complex mixture of three-dimensional non-peptide bonds would form.30 - The great majority of random chains of amino acids, even if optically pure, do not conveniently form complex secondary structures such as α helices, as assumed (figure 8).13 It is certainly true that alpha-helices of specific extant proteins do interact at precise portions of DNA; but this is neither coincidence nor a universal feature, and is caused by a precisely tailored set of spatial and electrostatic relationships, designed to serve a regulatory function.
- A large collection of mRNAs and tRNAs are needed at the same time and place. And these must provide or transmit the information to specify protein sequences! Sections of mRNAs must have exact sequences, and the complementary tRNAs to base-pair with them must already be available. Not only must the sequences be correct, their order with respect to each other must also be correct. And there must be a large number of such mRNAs, since many different proteins are needed. With a palette of only four nucleotides (nt) even a miniscule chain of 300 nucleotides offers 4300, or 4 × 10180 alternatives (ignoring all the structural isomers which could also form), the vast majority of which would be worthless. What natural process then, could have organized or programmed the mRNAs, and created the necessary tRNAs?
This is a fatal flaw in such models. The proportion of random polypeptides based on the 20 amino acids which are able to fold reliability to offer the chance of producing a useful protein is miniscule,31,32 to the order of one out of 1050. To provide the necessary information to generate one of the useful variants, something must organize the order of the bases (A,G,C and T) in the mRNAs. But nothing is available in nature which organizes the nucleotides into informationally meaningful sequences. - All the various peptides which need to be condensed together must be present. Where did these come from? Alberti writes, ‘Relatively short peptides (down at least to 17mers) recognize short specific sequences of double-stranded RNA or DNA.’33 The environment of the double strand chain offers far more useful physicochemical patterns to recognize than the single strand tRNA in the model, and even then, this would represent about one correct sequence out of 1022 (= 2017). Where did these peptides come from, and how was generation of the vast majority which are not desired avoided? Note that the necessary peptides would be of different lengths, depending on what needs to be recognized on a specific tRNA.
- Whether through ester bonds, weak hydrogen bonds or other interactions, without specific base-pairing as mediated by nucleotide polymers, all the countless varieties of polypeptides would not associate consistently at the same location on a tRNA-like molecule. For example, any free hydroxyl group of ribose is free to react with the carboxyl group of the peptide, forming an ester. All kinds of van der Waal or hydrogen bond interactions could also occur (figure 9). Therefore, the location of the peptide will not be reliably determined by any particular codon of the mRNA template.
- The mRNA-tRNA interaction alone is not reliable, requiring a considerable number of suitably located base-pairings between these strands, especially in the absence of any repair machinery, over long regions which is absurd. There will often be internal single-strand loops (figure 10), on the tRNA and mRNA. This will prevent a single codon on the mRNA from specifying uniquely and reliably the location of a putative polypeptide attached to the tRNA.
- It is important to understand what the author is calling ‘tRNA’ (see figure 8A).13 Key to his reasoning is that ‘Sequence-specific interactions between polypeptides and polynucleotides would result in the accumulation of specific polypeptide-polyribonucleotide pairs.’25 ‘Proximity between a peptide and an RNA molecule is likely to favour the formation of ester bonds between them.’25 The author assumes this results in the ancestral ‘tRNA’.
Each such ‘tRNA’ consists of a specific polypeptide sequence (and not single amino acids), which is chemically bonded to a unique single-strand RNA (figure 8B). Multiple tRNAs must then strongly base-pair to a matrix mRNA25 and be held rigidly25 at specific locations on the template mRNA. But a new peptide bond can only form between adjacent tRNAs if these are able to come into contact. This implies they must be attached at the ends of tRNAs, as shown in the original literature drawings,13 and that the tRNAs must be located close together on the mRNA. Otherwise the ester bond (between the peptide and RNA to form ‘tRNA’)25 would be buried and be inaccessible to the amino group of the second peptide it is to bond with. In figure 11 the carboxyl group of tRNA1 is shown inaccessible to the amino group of tRNA2.
To produce the ‘tRNAs’ the author assumes that portions of alpha-helices, each consisting of different series of amino acids to provide specificity, would ensure the unique interactions.25 However, random peptides can fold in an almost infinite number of ways and will not only form alpha-helices at specific locations (especially if racemic mixtures of amino acids are used). We must assume that polypeptide chains formed under natural conditions would be almost always amorphous polymers.
Perhaps there is an alternative to having to place proto-tRNAs very close together along the matrix mRNA. Suppose the locations where the tRNAs:mRNA base-pairings are more flexible, permitting them to eventually come close enough to react. This would happen when portions of tRNA.mRNA cannot base-pair, forming small bulges. Or if the tRNA would dissociate from mRNA and find itself in the vicinity of another tRNA it can react with. In other words, where the reacting ‘tRNAs’ are actually located with respect to the template mRNA would vary.
However, this would then destroy the notion of the ancient mRNA strand being a true coding template. It would not specify protein-sequences nor permit eliminating of tRNA-mRNA base-pair interactions (with the help of undefined ‘cofactors’ to hold tRNA and mRNA together) converging to the single codon used in the genetic code. - In this grand mixture of tRNAs and mRNAs what is to prevent their cross base-pairing? This would permit all the wrong kinds of peptides to be brought together where they could also polymerise.
- As peptide chains lengthen, they will start to fold into three-dimensional structures which would surround the esterized point of attachment with the tRNA. This would prevent for steric reasons other tRNAs from attaching in the area on the same mRNA, and the functional groups which are to react from approaching each other.
- Such a system has no means of self-replicating. Furthermore, postulating multiple covalent ester bonds implies some kind of hot, dry environment, which is inconsistent with the favoured evolutionary environments presented as candidates for where life would have arisen.
- Our greatest objection: nothing which needs to be explained has been seriously addressed. Precisely what are these ‘cofactors’ which are supposed to permit evolution to real ribosomes and aaRSs? These machines (ribosomes, aaRSs, etc.) require dozens of precisely crafted proteins, and it would take multiple miracles to generate precise molecular tools to systematically replace the base-pairings used to link the tRNA and mRNA strands,13 leaving only the codon-anticodon interactions. This is how modern ribosomes supposedly eventually arose. Note that in the earlier evolutionary stages a huge number of unique base-pairings were postulated, which permitted unambiguous association of each ancestral ‘tRNA’ with a precise portion of an mRNA. In the model, these base-pairings are systematically eliminated but the specificity (i.e. which tRNA attaches to which portion of an mRNA) must not be lost.
Concurrently, other undefined evolving ‘cofactors’ are responsible to eventually link a single amino acid to the correct tRNA, as modern aaRSs do. Is this feasible? According to the model,13 initially a multitude of different polypeptides (with 17 or more residues)13 each bonded to a specific RNA, leading to an ancient ‘tRNA’. (By ‘tRNA’ the author actually means an ancient ‘charged tRNA’ which uses a polypeptide and not a single amino acid). Twenty amino acids at seventeen positions leads to 2017 = 1.3 × 1022 possible ‘tRNAs’ plus many others having longer or shorter attached polypeptides. The carboxyl and amino ends of these large polypeptides then bond to form the primitive proteins (figure 11). The author does not explain how a tiny fraction of the more than 1022 alternatives were selected, nor does he consider whether a miniscule subset used would suffice to provide the minimal biological needs based on such crude proteins.
In the modern code, every residue of each protein is coded for, which permits any sequence of residues to be produced. The proposed ancient code, however, would only be able to code for individual large, discrete amino acid ‘blocks’.
Alberti believes that shorter and shorter polypeptide chains would eventually be needed to identify the correct RNA they must bond to. This process must culminate in true aaRSs, which charge a single amino acid to a specific RNA strand (i.e. real tRNAs). (Recall that initially longer polypeptides, which form alpha-helices, would be required to permit specific identification of the RNA they are to form an ester bond with). The author has provided no details which justify the claim that unguided nature could produce this effect with ‘cofactors’ or any other natural method.
But yet another fundamental point has been overlooked. It is assumed that originally discrete blocks of polypeptide bonded together, providing the necessary proteins. Amino acids are now being eliminated, leading to shorter ‘blocks’. As the polypeptides attached to the RNA strands shorten, different sequences would bond to the same RNA strand as before, producing an evolving code in which each ‘tRNA’ would be charged with different polypeptides. It is not obvious why modification of an individual ‘block’ by eliminating amino acids would still lead to acceptable primitive proteins. And evolving all the ‘blocks’ would lead to utter chaos. The exact same mRNA would now produce vastly different protein versions. - As cofactors are introduced between proto-tRNAs and mRNAs, and between peptides and tRNAs, the spatial relationships permitting earlier bonding of peptides together will be destroyed.



Instead of continuing with these kinds of vague chemical hypotheses, it seems more sensible for evolutionists to avail themselves of any chemical materials they wish (knowing full well they were of biological origin) and to show in a laboratory something specific and workable. If intelligently organizing all the components in any manner desired (besides simply reproducing an existing genetic system) can’t be made to work, then under natural conditions with > 99.999% contamination, UV light and almost infinite dilution, a code-based replicator is simply not going to arise.
(II) Coevolution of biosynthetically related amino acid pathways
In this view, the present code reflects a historical development. New, similar amino acids would evolve over time from existing synthesis pathways and be assigned to similar codons. Several researchers claim34 that biosynthetically related amino acids often have codons which differ by only a single nucleotide. It is also claimed35 that the class II synthetases are more ancient than class I, and so the ten amino acids served by class II would have arisen earlier in the development of the genetic code.
Objections. We cannot provide a thorough analysis of this hypothesis here. The argument is weakened considerably, however, by the fact that many amino acids are interconvertible. Even randomly generated codes show similar associations between amino acids which are biosynthetically related,34 and it is not at all clear which amino acids are to be considered biosynthetically related.36
Nature would have to experiment with many possible codes and have created many new biochemical networks to provide new amino acids to test. This would require novel genes. Nature cannot look ahead and sacrifice for the future, so each of the multitudes of intermediate exploratory steps cannot require deleterious stages. This poses impossible challenges to what chance plus natural selection could accomplish. We discussed the notion of testing different codes elsewhere.1
If only a subset of amino acids were used in an earlier life form, the necessary evidence should be available. The ‘highly conserved’ proteins, presumed to be of very ancient origin, should demonstrate a strong usage of the originally restricted amino acid set. This expectation is especially true if the extant sequences demonstrate little variability at the same residue positions. Furthermore, the first biosynthetic pathway could only have been built with proteins based on the amino acids available at that time. The residue compositions of members from both ancient and more modern pathways could be compared to see if a bias exists.
Is it unreasonable to demand this kind of supporting evidence? Suppose someone reported that the proteins used by the class II synthetases machinery relied on only the amino acids produced thereby. Every evolutionist alive would use this as final and conclusive proof for the theory. Then why should one be reluctant to make such a prediction? Without looking at the data yet, we predict this will not be the case.
(III) Evolution and optimisation to prevent errors
Some have proposed37–39 that genetic codes evolved either to minimize errors during translation of mRNA into protein, or the severity of the outcome40,41 which results. A similar proposal40,42 is that the effects of amino acid substitution through mutations are to be minimized by decreasing the chances of this occurring and the severity of the outcome should they occur. It would be desirable if random mutations would merely introduce residues with similar physicochemical properties.43,44
Amino acids can be characterized by at least 134 different physicochemical properties,45 begging the question as to which property or cluster of properties are most important. For example, measures of amino acid volumes seem less important than polarity criteria.46 In addition, C ↔ G mutations tend to be more frequent than A ↔ U mutations,47 for which an optimised genetic coding convention would need to take into account. Transition mutations48 tend to occur more frequently than transversion mutations.48 During translation (and DNA replication), transitional errors are most likely, since mistaking a purine for the other purine or a pyrimidine for the other one is, for stereochemical reasons, more likely.
Therefore, the best genetic codes would provide redundancy such that the most likely translation errors or mutations would result in the same amino acid very often. Freeland and Hurst49 took this into account when comparing with a computer a million randomly generated codes having the same pattern of codon assignments to different amino acids as the standard code. Using a measure of hydrophobicity as the only key attribute to be protected by a coding convention (and taking nucleotide mutational bias into account) they found only one code out of a million which by the hydrophobicity criterion alone, would be better. We are convinced that taking more factors to be optimised into account would reveal this proportion to be much smaller.
Hydrophobicity reflects the tendency of amino acids to avoid contact with water and to be present in the buried inner core of folded proteins. Unfortunately, no best measure of hydrophobicity for amino acids has been agreed upon, and at least 43 different laboratory test methods have been suggested.50 The different criteria often lead to very different ranking of amino acid hydrophobicity.50
Others have thought that mutability played an important role: robustness was important for conservation of some proteins but mutability was required to permit evolution also.51 Still others have focused on overall effects of mutations on protein surface interactions with solvent52 which lead to protein secondary features such as alpha helices and beta sheets.53
Having the option of using different codons to code for the same amino acid can be advantageous. For example, if a low concentration of the protein is desired, synonymous codons can be used which lead to slower translation54,55 by taking advantage of the fact that the corresponding aaRSs are often present in very different proportions. If a specific tRNA is only present in a low concentration, the target codon must wait much longer to be translated than if the tRNA is highly available. Sharp et al. reported56 that highly-expressed genes indeed preferentially use those codons which lead to faster translation. This is realized by maintaining different concentrations of the corresponding aaRSs. Translation of an mRNA can be slowed down if a rare codon being translated by a ribosome needs to wait until the appropriate charged tRNA stumbles into that location, for example to give time for a portion already translated to initiate folding.57
It is not obvious which property or properties of amino acids should be conserved in the presence of mutations. One suggestion by Freeland and colleagues16 is to use point accepted mutations (PAM) 74–100 matrix data. Comparing aligned versions of genes which have been mutating (in organisms presumably sharing a common ancestor) after about 100 million years would presumably reveal which amino acid substitutions are more variable or on the other hand, more intolerant to substitution. The authors then examined whether the assignment of synonymous codons protected against such changes, and concluded58 the universal genetic code ‘achieves between 96% and 100% optimisation relative to the best possible code configuration’.
Mechanisms for codon swapping. There are various scenarios1 as to how codons could begin to code for a different amino acid. According to the Osawa-Jukes model59 mutations cause some codons to disappear from the genome, and the relevant tRNA genes, being superfluous, disappear. At this point these genomes would not have all 64 of the possible codons present in protein-coding regions. This process is thought to be caused by a mutational bias leading to higher A-T or G-C genome content. When later this mutational bias reverses, the missing codons would begin to show up somewhere in the genome. These could no longer by translated, since the corresponding tRNA is lacking. But duplication of a gene for tRNA followed by mutations at the anticodon position might permit recognition of the new codon on the mRNAs, which would now translate for a different amino acid.
The Schultz-Yarus60 model is similar but permits the codon to remain partially present in the genome. Mutations on a duplicated tRNA produces a different anticodon or a new amino acid charging specificity and thereby ambiguous translation of a codon (i.e. the same codon could be identified by different tRNAs). Natural selection would then optimise a particular combination. Incidentally, in some Candida species CUG will encode either serine or leucine,60 depending on the circumstances.
Objections. We have discussed various difficulties with the notion of trial-and-error attempts to find better coding conventions elsewhere.1 There are over 1.5 × 1084 codes which could map 64 codons to 20 amino acids plus at least one stop signal.1 This is a huge search space, and most of the alternatives would have to be rejected. But when would nature ‘know’ a better or worse coding convention is being explored? Several stages are needed.
- Many genes would have to be functionally close to optimal so that natural selection could identify when random mutations would produce inferior versions. This means that an unfathomably large number of mutational trials would be needed to produce many optimal genes. Interference with a mutating genetic code would hinder natural selection’s efforts.
- One or more codons would have to be recoded and the effects throughout the whole genome ascertained. During this process many codons would be ambiguous, such that a myriad of protein variants would be generated by almost all genes, in the same individual. Natural selection would be faced with a continuously changing evaluation as to whether the evolving codon would be advantageous.
- One evolving coding convention needs to be completed, before another one can be initiated. For example, if during the interval when 70% of the time a codon leads to amino acid ‘a’ and 30% of the time to ‘b’ additional codons were to also become ambiguous, cellular chaos would result. Besides, we see nowhere in nature examples of a multitude of ambiguous codons present simultaneously in an organism.
Generating a new code demands removing the means of producing the original coding option. Depending on the mechanism of code evolution, this could mean removing duplicate tRNA or aaRS variants throughout the whole population. This is going to be near impossible since the selective advantage would be minimal, and at best would consume a huge amount of a key evolutionary resource, time.
Nature can’t know in advance which coding convention would eventually be an improvement. An initial 0.1% ambiguity in a single codon, which may be limited to a single gene (such as the case of specific chemical modifications of mRNA), is hardly going to be recognized by natural selection. Note that this 0.1% alternative amino acid would be distributed randomly across all copies of this codon on a gene, and the resulting proteins would be present in multiple copies. The alternative residue would be present in only a small minority of these proteins, and randomly.
Once a new code has been fixed, this limits the direction future evolutionary attempts can take. There is no mechanism in place to allow a return to a previous code once it was abandoned other than to re-evolve back to that system. Given the large number of unrelated factors which determine prokaryote survival from the external environment and quality of the genetic system, natural selection would not be provided with any consistent guidance. The rules would change constantly. And a multitude of criteria need to be taken into account simultaneously in deciding what to do with each codon. Codons can be used by several codes not related to specifying amino acids,61 and the relative importance of the tradeoffs will change constantly.
Discussion
We believe the genetic apparatus was designed, and agree there must be a logical reason for the codon → amino acid mapping chosen. We suspect that protection against the effects of mutations is indeed one of the factors which went into the choice made. This would require foreknowledge of all the kinds of genes needed by all organisms and a weighting of the damage each kind of amino acid substitution could cause. Optimal design may also require variants of the code to be used for some of the intended organisms. But we wish to emphasize that the code to determine amino acid order in proteins is not the whole story. Many other codes61–63 are superimposed on the same genes and noncoding regions, and must also be taken into account in the design of the code. Various nucleotide patterns are used for DNA regulatory and structural purposes. DNA must provide information for many other processes besides specifying protein sequences. These requirements affect which code would be universally optimal.
Interestingly, a design theoretician may well make a similar suggestion to that of Freeland and colleagues16 mentioned above, but based on other reasoning. To a first approximation, the optimal design of the same proteins in different organisms would be similar. For various reasons, occasionally substituting an amino acid would be better. For example, in hot environments the proteins may have to fold more tightly, whereas this design could prevent enzymatic activity under cooler conditions, by embedding a reactive site too deeply in a rigid hydrophobic code. In general, optimal protein variants must often use residues with similar properties, such as hydrophobicity or size, at a given position. The genes would not be similar due to common descent but by design requirements. Mutations would subsequently generate less than optimal variants which would still be good enough.
An intelligently planned genetic code would have taken this into account. Therefore, to a first approximation, comparing aligned genes and determining substitutability patterns would indeed provide useful information as to amino acid requirements and use of alternatives. If enough taxa living in many environments are used as a dataset, we should be able to obtain a good idea as to the amount of variability homologous proteins would have. Of course ‘noise’, in the form of random mutations, will also be present. Knowledge of other superimposed codes not responsible for coding for protein sequences, would permit even better quantification as to how optimal the standard code really is. Various alternative codes must satisfy many design requirements, and the optimal one will do best for all demands placed on it.
There is however one key difference in the reasoning. We propose that God knew what the ideal protein sequences should be, and therefore which needed protection from mutations, and all the other roles nucleotide sequences need to play. The evolutionist here has a problem. Fine-tuning hundreds or thousands of genes concurrently via natural selection to produce a near optimal ensemble is absurd. During the time when the regulation of biochemical networks and enzymes are being optimised, the rules in the form of the code would also be changing. Yet a Last Universal Common Ancestor (LUCA) supposedly already had thousands of genes64 and the full set of tRNA synthetases and tRNAs7 about 2.5 billion years ago.65 Actually, other lines of reasoning66 have led to the belief that the genetic code is almost as old as our planet. In other words, it had virtually no time to evolve and yet is near optimal in the face of over 1.5 × 1084 alternative 64 → 21 coding conventions.
We see evidence everywhere of cellular machinery designed to identify, ameliorate and correct errors. In sexually reproducing we observe that genes are duplicated, which mitigate the effects of many deleterious mutations and thereby help organisms retain morphologic function. Many evolutionists now propose nature has attempted to conserve complex functionality from degradation. All this implies that a highly optimal state has been achieved which nature is trying to retain. More consistent with evolutionary thought would be proposals which encourage ‘evolvability’ or adaptation. Evolution from simple to specified complexity is not achieved by hindering change.
Summary
A key element in evolutionary theory is that life has gone from simple to complex. But requiring the minimal components of a genetic code to be simultaneously in place without intelligent guidance is indistinguishable from demanding a miracle. No empirical evidence motivated searches for simpler or less optimal primitive genetic codes. Once the possibility of Divine activity has been excluded as the causal factor, an almost unquestioning willingness to accept absurd notions is created among many scientists. After all, it must have happened!
We conclude that no one has proposed a workable naturalistic model that shows how a genetic code could evolve from a simpler into a more complex version.
References
- Truman, R. and Terborg, P., Genetic code optimisation: Part 2, J. Creation 21(3):84–92, 2007. Return to text.
- Judson, O.P. and Haydon, D, The genetic code: what is it good for? An analysis of the effects of selection pressures on genetic codes, J. Mol. Evol. 49:539–550, 1999. Return to text.
- Trevors, J.T. and Abel, D.L., Chance and necessity do not explain the origin of life, Cell Biology International 28:729–739, 2004. Return to text.
- There are a few minor variants which are believed to be degenerations of the universal code. See Osawa S., Evolution of The Genetic Code, Oxford University Press, Oxford, 1995. Return to text.
- Trevors and Abel, ref. 3, p. 729. Return to text.
- See www.us.net/life/. The site, in its ‘Description and Purpose of the prize’, states, ‘“The Origin-of-Life Prize” ® (hereafter called “the Prize”) will be awarded for proposing a highly plausible mechanism for the spontaneous rise of genetic instructions in nature sufficient to give rise to life. To win, the explanation must be consistent with empirical biochemical, kinetic, and thermodynamic concepts as further delineated herein, and be published in a well-respected, peer-reviewed science journal(s).’
‘The one-time Prize will be paid to the winner(s) as a twenty-year annuity in hopes of discouraging theorists’ immediate retirement from productive careers. The annuity consists of $50,000.00 (U.S.) per year for twenty consecutive years, totalling one million dollars in payments.’ Return to text.
- Knight, R.D., Freeland, S.J. and Landweber, L.F., Selection, history and chemistry: the three faces of the genetic code, TIBS 24:241–247, 1999. Return to text.
- Woese, C.R., The Genetic Code, Harper & Row, New York, 1967. Return to text.
- Pelc, S.R. and Welton, M.G.E., Stereochemical relationship between coding triplets and amino-acids, Nature 209:868–872, 1966. Return to text.
- Dunnill, P., Triplet nucleotide-amino acid pairing: A stereochemical basis for the division between protein and nonprotein amino acids, Nature 210:1267–1268, 1966. Return to text.
- Root-Bernstein, R.S., Amino acid pairing, J. Theor. Biol. 94:885–904, 1982. Return to text.
- Hendry, L.B. and Whitham, F.H., Stereochemical recognition in nucleic acid-amino acid interactions and its implications in biological coding: a model approach, Perspect. Biol. Med. 22:333–345, 1979. Return to text.
- Alberti, S, Evolution of the genetic code, protein synthesis and nuclei acid replication, MLS, Cell. Mol. Life Sci. 56:85–93, 1999. Return to text.
- Cedergren, R. and Miramontes, P., The puzzling origin of the genetic code, Trends Biochem. Sci. 21:199–200, 1996. Return to text.
- Freeland, S.J., Wu, T. and Keulmann, N., The Case for an Error Minimizing Standard Genetic Code, Origins of Life and Evolution of the Biosphere 33:457–477, 2003. p. 458: ‘Under this model, selection operates independently from the previous evolutionary forces, potentially overwriting the footprints of stereochemical origins and biosynthetically mediated code expansion.’ Return to text.
- Freeland, S.J., Knight, R.D., Landweber, L.F. and Hurst, L.D., Early fixation of an optimal genetic code, Mol. Biol. Evol. 17(4):511–518, 2000. Return to text.
- Trifonov, E.N., Consensus temporal order of amino acids and evolution of the triplet code, Gene 261:139–151, 2000. See overview and references on p. 141. Return to text.
- Bergman, J., Why the Miller-Urey research argues against abiogenesis, J. Creation 18(2):74–84, 2002. Return to text.
- Miller, S.L., Production of amino acids under possible primitive earth conditions, Science 117:528–529, 1953. Return to text.
- Truman, R. and Terborg, P., Why the shared mutations in the hominidae exon X GULO pseudogene are not evidence for common descent, J. Creation 21(3):118–127, 2007. Return to text.
- Simpson, S., Life’s first scalding steps, Science News 155(2):24–26, 1999; p. 26. Return to text.
- Flowers, C., A Science Odyssey: 100 Years of Discovery, William Morrow and Company, New York, p. 173, 1998. Return to text.
- Shapiro, R., Origins; A Skeptics Guide to the Creation of Life on earth, Summit Books, New York, p. 99, 1986. Return to text.
- Campbell, N.A., Biology, Benjamin/Cummings, Redwood City, CA, 1993. Return to text.
- Alberti, ref. 13 p. 87. Return to text.
- Sarfati, J., Origin of life: the chirality problem, J. Creation 12(3):263–266, 1998. Return to text.
- Joyce, G.F. and Orgel, L.E., Prospects for understanding the origin of the RNA world; in: Gesteland, R.F., Cech, T.R., Atkins, J.F. (Eds.), The RNA world, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY; pp. 49–78, 1999. Return to text.
- Sarfati, J., Hydrothermal origin of life? J. Creation 13(2):5–6, 1999. Return to text.
- Imai, E., Honda, H., Hatori, K., Brack, A. and Matsuno, K., Elongation of oligopeptides in a simulated submarine hydrothermal system, Science 283(5403):831–833, 1999. Return to text.
- Thaxton, C.B., Bradley, W.L. and Olsen, R.L., The Mystery of Life’s Origin, Lewis and Stanley, Dallas, TX, 1992. See chapter 4. Return to text.
- Truman, R. and Heisig, M., Protein families: chance or design? J. Creation 15(3):115–127, 2001. Return to text.
- Truman, R., Searching for needles in a haystack, J. Creation 20(2):90–99, 2006. Return to text.
- Alberti, ref. 13, p. 90. Return to text.
- Knight et al., ref. 7, p. 243. Return to text.
- Hartman, H., Speculations on the origin of the genetic code, J. Mol. Evol. 40:541–544, 1995. Return to text.
- Amirnovin, R., An analysis of the metabolic theory of the origin of the genetic code, J. Mol. Evol. 44:473–476, 1997. Return to text.
- Woese, C.R., On the evolution of the genetic code, Proc. Natl Acad. Sci. USA 54:1546–1552, 1995. Return to text.
- Goldberg , A.L. and Wittes, R.W., Genetic code: aspects of organization, Science 153:420–424, 1966. Return to text.
- Woese, C.R. The Genetic Code: The Molecular Basis for Genetic Expression, Harper and Row, 1967. Return to text.
- Sonneborn, T.M, Degeneracy of the genetic code: extent, nature, and genetic implications; in: Bryson V., Vogel H.J. (Eds.), Evolving genes and proteins, Academic Press, New York, NY, 1965. Return to text.
- Ardell, D.H., On error minimization in a sequential origin of the standard genetic code, J. Mol. Evol. 47:1–13, 1998. Return to text.
- Epstein, C.J., Role of the amino-acid ‘code’ and of selection for conformation in the evolution of proteins, Nature 210:25–28, 1966. Return to text.
- Wolfenden, R.V., Cullis, P.M. and Southgate C.C.F., Water, protein folding, and the genetic code, Science 206:575–577, 1979. Return to text.
- Haig, D. and Hurst L.D., A quantitative measure of error minimization in the genetic code, J. Mol. Evol. 33:412–417, 1991. Return to text.
- Sneath, P.H.A., Relations between chemical structure and biological activity in peptides, J. Theor. Biol 12:157–195, 1966. Return to text.
- Ardell, ref. 41, p. 2. Return to text.
- Ardell, ref. 41, p. 8. Return to text.
- In transition mutations a pyrimidine is substitute by another pyrimidine, or a purine by a purine (A ↔ G or C ↔ T). In transversion mutations a pyrimidine is substitute by a purine or vice-versa (A ↔ C, G ↔ T, G ↔ C or A ↔ T). Here A, C, G and T represent the four possible nucleotides used by DNA. Return to text.
- Freeland, S.J. and Hurst, L.D., The genetic code is one in a million, J. Mol. Evol. 47:238–248, 1998. Return to text.
- Trinquier, G. and Sanejouand,Y.-H., Which effective property of amino acids is best preserved by the genetic code? Protein Engineering 11(3):153–169, 1998. Return to text.
- Maeshiro, T. and Kimura M., The role of robustness and changeability on the origin and evolution of genetic codes, Proc. Natl Acad. Sci. USA 95:5088–5093, 1998. Return to text.
- Sitaramam, V., Genetic code preferentially conserves long-range interactions among the amino acids, FEBS Lett. 247:46–50, 1989. Return to text.
- Dufton, M.J., Genetic code redundancy and the evolutionary stability of protein secondary structure, J. Theor. Biol. 116:343–348, 1985. Return to text.
- Grosjean H. and Fiers W, Preferential codon usage in prokaryotic genes: the optimal codon–anticodon interaction energy and the selective codon usage in efficiently expressed genes, Gene 18:199–209, 1982. Return to text.
- Sørensen, M.A. and Pedersen, S. Absolute in vivo translation rates of individual codons in Escherichia coli—the two glutamic acid codons GAA and GAG are translated with a threefold difference in rate, J. Mol. Biol. 222:265–280, 1991. Return to text.
- Sharp, P.M., Stenico, M., Peden, J.F. and Loyd, A.T., Codon usage: mutational bias, translational selection or both? Biochem. Soc. Trans. 21:835–841, 1993. Return to text.
- Kimchi-Sarfaty, C. et al., A ‘silent’ polymorphism in the MDR1 gene changes substrate specificity, Science 315:525–528, 6 Jan. 2007; published online Dec. 21 2006; www.sciencemag.org. Return to text.
- Freeland et al., ref. 16, p. 515. Return to text.
- Osawa, S. and Jukes, T.H., Evolution of the genetic code as affected by anticodon content, Trends Genet. 4:191–198, 1988. Return to text.
- Yarus, M. and Schultz, D.W., Toward an explanation of malleability in genetic coding, J. Mol. Evol. 45:1–8, 1997. Return to text.
- Trifonov, E.N., Genetic sequences as product of compression by inclusive superposition of many codes, Molecular Biology 31(4):647–654, 1997. Return to text.
- Segal E., Fondufe-Mittendorf Y., Chen L., Thastrom A., Field Y., Moore, I.K., Wang, J.P. and Widom J., A genomic code for nucleosome positioning, Nature 442(7104):772–778, 2006. Return to text.
- Wade N., Scientists Say They’ve Found a Code Beyond Genetics in DNA, The New York Times, 25 Jul. 2006. Return to text.
- Ouzounis, C.A., Kunin, V., Darzentas, N. and Goldovsky L., A minimal estimate for the gene content of the last universal common ancestor: exobiology from a terrestrial perspective, Res. Microbiol. 157:57–68, 2006. Return to text.
- Gu., X, The age of the common ancestor of eukaryotes and prokaryotes: statistical inferences, Mol. Biol. Evol. 14(8):861–866, 1997. Return to text.
- Eigen, M., Lindemann, B.F., Tietze, M., Winkler Oswatitsch, R., Dress, A. and Haeseler, A., How old is the genetic code? Statistical geometry of tRNA provides an answer, Science 244:673–679, 1989. Return to text.
Readers’ comments
Comments are automatically closed 14 days after publication.