

Journal of Creation 19(3):116–127, December 2005
Browse our latest digital issue Subscribe
The ubiquitin protein: chance or design?
A dataset based on all known functional sequences of ubiquitin (UB) has been prepared. This confirmed the extraordinary intolerance of this protein to substitution of alternative amino acids. Probabilistic calculations suggest evolutionary processes did not produce such unusual sequences.
It was discovered that three amino acid positions on the chain are distinctive for organisms belonging to the animal, plant and fungus kingdoms. A fourth variant has already been reported. The unexpected identification of three new distinct classes of UB has implications for evolutionary theory. It requires the common ancestor for all UB families to be essentially simultaneous with the origin of the first eukaryote.
Ubiquitin (UB) is a key protein believed to be present in all eukaryote type cells. This means that organisms such as yeast, plants, fish, mammals, and so on, all possess UB. Evolutionary theory explains this by postulating a common ancestor. However, the narrow range of amino acid sequence variability found throughout nature seems inconsistent with the notion of an ancient origin and very little change through mutations. Given the large amount of data available in public databases, I decided to see how much variety in amino acid sequence exists and how the data might be interpreted from a creationist or evolutionist framework.
Some classes of proteins show considerable variety in the sequences of amino acids, and do not differ too greatly from other families of proteins. Whether the creationist or evolutionist viewpoint is correct is not so easy to argue in these cases. But extreme examples might be found which are difficult to explain in evolutionary terms. Such would be the case for a narrow range of acceptable variability over a wide range of taxa and absence of other protein families with similar sequences.
Billions of years of mutations in genetically separate lineages would surely produce much change. If very little variety is found, then surely few alternatives are functional. And if virtually none are functional, an evolutionary starting point for natural selection to act upon would be extraordinarily unlikely to be found by chance mutations. An absence of similar classes of proteins would suggest the lack of intermediate ‘stepping stones’ linking useless genes with those coding for a highly specialized proteins.
Ubiquitin is an interesting candidate to examine, since the standard university textbooks claim there is very little variability in sequence across many kinds of organisms. The exact amount has not yet been reported in the literature, using all data now available. No effort to identify candidate evolutionary ancestral protein classes appear to have been reported either.
There is a final issue, which in the case of ubiquitin will be dealt with later. Are there examples of highly constrained proteins for which a large number of other proteins are required simultaneously, before they can offer any kind of selective advantage? The existence of such molecular machines would suggest ‘Irreducible Complexity’: natural selection could only be effective once everything is in place, but highly unlikely components are also required.
Dataset of sequences
A blast-p1 search based on a known ubiquitin sequence identified about 860 similar sequences, which were downloaded to a PC.
Some pre-processing was necessary before one can examine the data in more detail. A file was created after excluding:
- Polypeptide sequences attached to the ubiquitin protein. I was interested at this time in the functionality and variability of the free UB
- Sequences obtained from viruses. I could not determine the source organism these had been picked up from and whether they are functional
- Exotic sequences found in unicellular parasites. I could not be certain these UBs function normally, given that the UB mRNA is presumably also available via the host
- Duplicate sequences from the same organisms. (Often UB is present in many copies on the same genome)
- Sequences for which the organism was not annotated in the source database.
Dataset cleanup was done manually to ensure that no data would be lost. For example, when identical sequences for the same organism was found, reported from different laboratories, the first one in the data file was kept. In the case when a large number of UBs were fused together, the first one was retained. Sequences from viruses were removed and saved in a separate file.
Scripts programmed with ActiveState perl2 confirmed the integrity of the effort. Scripts such as shown in Appendix A3 and Appendix B4 can save about a week of effort in these kinds of activities. New data added to the public databases1 later may require future datasets to be generated, and such scripts can save much effort.
Sequence alignment

Table 1. Amino acid abbreviations and chemical structures. (Click on picture to see a larger image)
The sequences in the dataset were examined after optimally aligning the residues with the ClustalX version 1.8.1 utility 4 (Fig. 15). The final data is also provided online6 and consists of data from 158 different organisms. For the meaning of one-letter amino acid codes see Table 1. Unique patterns can be ascertained for all the ubiquitins, such as the terminal RGG (see table 1 for code) residues, which can be used as parameters in a perl script (Appendix A)3 to extract and organize sequences. In Table 2 (available online7), the number of alternative amino acids found at each of the 76 residue positions of ubiquitin is shown based on a table (available online8) and Table 3 documents the residues in which some variability was found. In 46 positions the exact same amino acid was found, meaning that about 60% of UB seems to tolerate no mutations at all and in 17 other positions a single alternate amino acid was occasionally found (Table 4). In almost all the latter cases this exception was found in only a single organism and some of these may simply be incorrectly reported data.
Probability analysis
Before natural selection could begin fine-tuning UB, a protein with a suitable sequence providing minimal functionally must be available. A novel gene must code for a polypeptide. There are (20)n alternative polypeptide sequences, where n is the length of the chain. What is the probability a random polypeptide chain 76 amino acids long might provide an evolutionary starting point?
Information Theory calculations
Probability calculations of this kind based upon Information Theory were discussed previously.9 The method excludes ‘low probability sequences’, such as chains which consist of many amino acids which are coded for by only a single codon. Among random DNA chains I argued earlier9 that such sequences are extremely unlikely and information theory takes such statistical factors into account.
The method assumes that the presence of a particular amino acid at a given residue position does not change the probability of others being present elsewhere. This is known to be incorrect. Different amino acids may be found in various functional UBs at various residue positions. This does not imply that all these mutations would be acceptable when present concurrently in the same UB. This erroneous extrapolation would suggest 16,796,1600 acceptable alternatives10 exist using the data in Table 3 based on 55 alternatives actually reported, but no more than 15,820 expected alternatives would be justified by the most generous assumptions.10 The proportion estimated to be functional by information theory, by neglecting the effect of context dependence, is therefore too high. However, this does compensate for the fact that not all conceivable acceptable mutations have been identified in my ubiquitin dataset.
The calculations shown in Appendix 1 suggest that a proportion of about 4X10-83 polypeptides 76 residues long would produce a functional UB in living organisms.

Table 3. Ubiquitin sequences with some variability, based on online data. (Click on picture to see a larger version)

Table 4. Amino acid residue position on ubiquitin showing no variability or at most a single alternative amino acid for only one organism. Based on ref.8. (Click on picture to see a larger version)
Different families of ubiquitin
Examination of the sequences at the locations where variability is present reveals that there are actually different families of UB, grouped by the animal, plant and fungus kingdoms. Residue positions 19, 24 and 57 are especially diagnostic, and to a minor extent also positions 16 and 28.
The animal PES pattern
The ‘PES’ amino acid pattern shown in Table 5 holds for mammals, fish, insects, amphibians, many protozoa, etc. From this table one notes that UB for human and oyster sequences are identical.11 UB from protozoa such as Leishmania tarentolae and Leishmania major are identical to each other and differ from the human UB at only 2 residues.12 Not many UB sequences are available for micro-organisms, and those examined are typically pathogens of medical interest.
The PES pattern is so consistent, that an exception was quickly noticed in the case of two worms.13 Further investigation and correspondence with the main author of the papers clarified that these are parasite worms which feed on plants. The UB pattern they possess are indeed characteristic of plants, and are fused to polypeptides the worms inject into a host plant to modify its developmental behaviour. Presumably these worms also possess UB variants with the expected PES pattern, as found in other worms (see the online database, (ref. 5).

Table 5. The distinctive PES EA and minor AES EA pattern for animal-like organisms. (Based on amino acids found at residue positions 19, 24, 57, 16, 28). See ref. 5. (Click on picture to see a larger version)
The minor AES pattern
A few organisms had A (alanine) instead of P (proline) at residue position 19: some worms, bees, one cockroach, three ciliated protozoans and one amoeba. The possibility of errors in recording needs to be checked, especially where only a single sequence is available. In the case of some worms and bees, multiple reports confirm that this alternative amino acid is present. If the original created organisms had the PES pattern, a single mutation from any of the four codons for proline (C CU, C CC, C CA, C CG) could code now for alanine (G CU, G CC, G CA, G CG).

Table 6. The distinctive SDA EA pattern for plant-like organisms. (Based on amino acids found at residue positions 19, 24, 57, 16, 28). See ref. 5. (Click on picture to see a larger version)
The plant SDA pattern
The ‘SDA’ pattern shown in Table 6 covers a wide range of plants.
The fungus SDS pattern
The ‘SDS’ pattern shown in Table 7 characterizes fungi (which includes yeast). The lack of variability within very different fungi is dramatic. For example, Magnaporthe grisea (rice blast fungus) and Tuber borchii (an edible truffle / mushroom) ubiquitins are 100% identical.14
Miscellaneous patterns
The UB sequences of some organisms do not fall into one of the categories displayed so far (Table 8). One red algae showed the animal PES pattern. Perhaps it was contaminated, for example with UB from coral. Only one sequence was found for this organism. A pattern ‘SEA’ was found for green algae, gliding flagellates and an amoeba. The plant pattern SDA EA is similar to SEA especially when we consider that the amino acids D (aspartic acid) and E (glutamic acid) are very similar. Although plants always showed EA in residue positions 16 and 28 (Table 6), greater variability was found in the few exceptional organisms shown in Table 8. Supplemental sequences are needed to determine if there are additional patterns to those identified for animals, plants and fungi.
Another category of ubiquitin
One or two amino acids separate chains of otherwise continuous UB in members of the Cercozoa and Foraminifera protist lineages (Table 9). Since gene expression begins with a methionine (M), this implies that the protein will have one or two extra amino acids added on. This is very interesting, since the end glycine’s (G76) carboxyl (COOH) group plays a critical role: it is used in a precisely engineered manner with the help of several enzymes to tag a target protein. This forms an isopeptide bond with an ε-amino group of a lysine. But amino acids following G76 would prevent conjugation by bonding to this key carboxyl group. It is very likely special enzymes not present in other organisms remove the extra residues.
Some of the examples in Table 9 show a dipeptide (i.e. the amino acids MS) preceding the usual M used to initiate gene expression. Either this is also cut out, or the longer ubiquitin variant is also functional. The authors15 considered these UB features significant enough to claim close evolutionary kinship between Cercozoa and Foraminifera, although analysis of numerous gene sequences created hopelessly contradictory phylogenetic trees. The evidence supports the proposal that there are at least four isolated families of UB proteins. From Table 9 one observes that most of the organisms would be classified by their morphology as plant-like and usually display a pattern SEA.
Discussion
Evolutionary probabilities
We need to see whether the chances of obtaining a functional UB from among random 76 amino acid polypeptide chains, 4X10-83, for natural selection to act upon, can be met by natural processes. An alternative model, in which UB might have arisen from a pre-existing gene, will be explored in another paper. In two independent papers9,16 about the same estimate for maximum number of organisms which could have lived on earth during 4 thousand million years was reported: 1046. Estimates of mutational rates vary between 10-7 and 10-12 per nucleotide17-21 according to organism. For small bacteria-like genomes this implies most of these organisms had no mutations between most generations. Clearly the odds of producing just one UB gene (plus the necessary de-ubiquitinating enzyme needed to free it from the rest of the polypeptide) by random mutations are infinitesimally small. Much greater mutational rates on average would lead to error catastrophe as deleterious errors accumulate over all members of the population.
The metabolic cost for expressing a duplicated gene in yeast has been recently reported22 and the author confirmed that a negative selection coefficient would result. Rapidly reproducing, small organisms, which express genes with no biological value, would be selectively disadvantaged and the lineage sizes streamlined after just a few years. The resulting absence of candidate genetic building material in such putative ancient, primitive organisms, to permit generating novel genes precludes the possibility of generating new, complex biological functions through evolutionary mechanisms.
In addition, non-coding sequences necessary to express worthless genes would be quickly deactivated by random mutations, such that even if a useful DNA sequence were to arise by mutational chance, the protein would not be generated.
These kinds of probability considerations pose severe credibility challenges to evolutionary theory at three places:
- The origin of a primitive genetic system related to the extant ones (based on 64 codons, 20 amino acids and a ‘stop’ instruction) would require dozens of unrelated proteins ab initio.
- Increase in biological functionality, to explain what we observe today, requires thousands of new kinds of proteins/genes to be generated
- The total number of evolutionary trials, based on ca. 1046 total organisms, must cover all destructive and neutral mutations in the earth’s history; and still leave enough trials to initiate and then fine-tune thousands of useful novel proteins by Darwinian mechanisms.
It is apparent that trial-and-error, using all available organisms that might ever have lived on earth according to the evolutionist framework, could not be expected to generate a single minimally functional UB gene. I believe argument (iii) has not been developed in the literature yet and I intend to explain it later. Creationists and agnostics have argued that available time constrains the number of evolutionary trials. The constraint in (iii) claims that the total number of mutants that have lived is insufficient to explain the sum total of all genes known to exist presently.23
Probabilities in small jumps
In arguing that the odds of ever generating an initial gene coding for UB are essentially zero, I have implicitly assumed that a random sequence was the starting point.

The current evolutionary thinking is that new functions usually arose from a gene duplication event followed by subsequent divergence via mutations. Suppose other genes coding for proteins similar to ubiquitin were found on a genome. This is illustrated conceptually in Fig. 2. The key idea is that some of the mutants of two genes may be much more similar than the typical or consensus sequences. A few fortunate mutations might provide an evolutionary initial point much more easily than starting from a random sequence. Might not the statistical challenge become considerably lower?
The plausibility of the concept would need to be examined on a case-by-case basis. Several conditions must be met.
- The duplicated gene must not interfere with the expression of other genes.
- The diverging proteins and mRNAs must not interfere with each other. Given the sequence similarity, interference is very likely.
- All other components of the new function must already by present if positive selection is to occur.
- The new lineage must survive genetic drift and the mentioned selective disadvantage.
To generate the original ubiquitin as illustrated in Fig. 2, another similar gene must in fact have existed. The only candidates available in existing sequence databases are so-called ubiquitin-like (UBL) proteins. In a follow up paper, I’ll argue that evolving ubiquitin from a chain of preceding UBLs is actually more difficult than from a random stretch of DNA.
The very limited variability among known ubiquitin sequences (Table 4) implies that the cluster of alternatives near the presumably near-optimal consensus sequence (Fig. 2) is very narrow. This suggests that UB homologs would have to be first generated via gene duplication and then many amino acids mutated, producing non-functional UB proteins, before a minimally useful new gene could evolve. Requiring more than five amino acids to be modified for this purpose is statistically unrealistic.24 The sequences of several kinds of UBLs display very limited variability across organisms, and are too far removed from UB sequences to have derived from a common ancestor.25
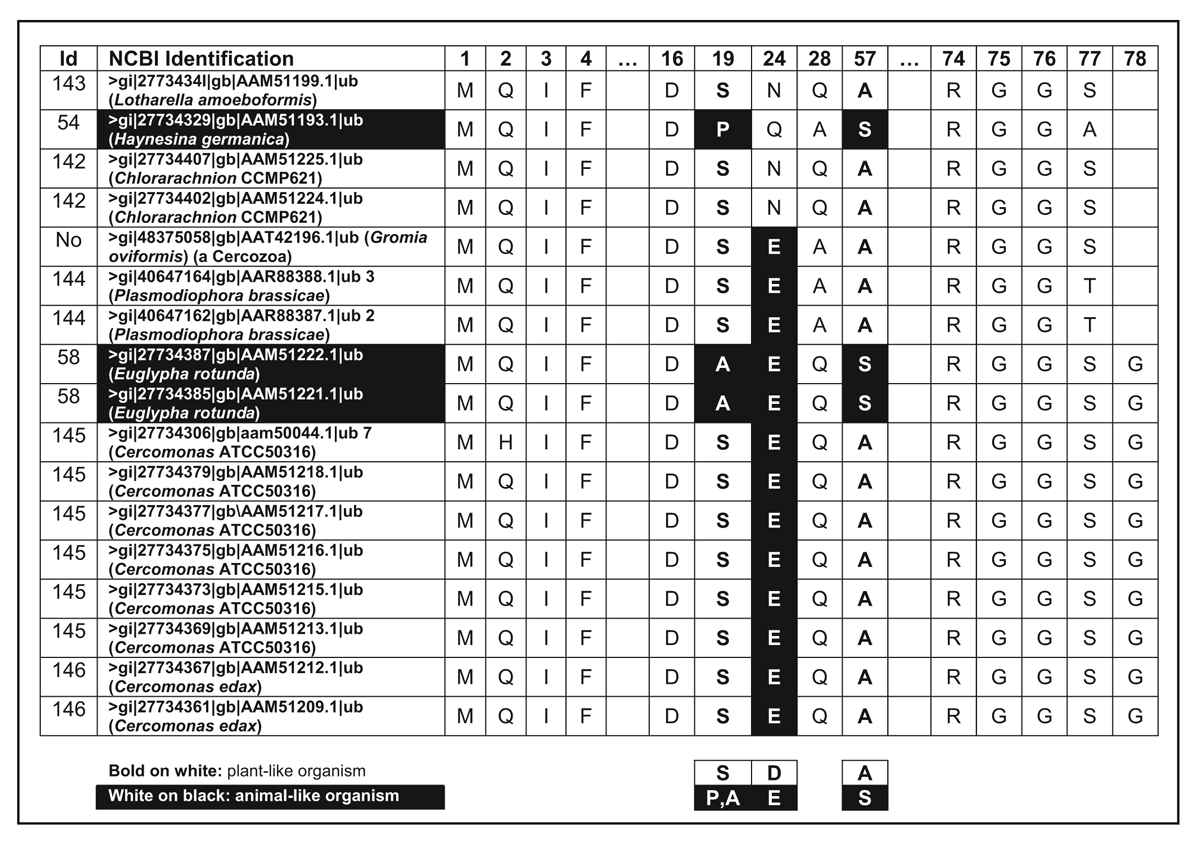
Table 9. Polyubiquitin sequences separated by one or two amino acids. (Click on picture to see a larger version)
Ubiquitin biochemistry
So far I have concentrated on the improbability of producing the UB protein only. But UB alone is biologically worthless, like virtually all other proteins. It is only useful when integrated into a process with other bio-chemicals. For UB to have selective value we need to look at the whole picture.
The 76-residue UB is rarely encoded by just a single gene,26,27 but is part of larger proteins, as illustrated in the standard Fasta format sequences downloaded (Fig. 3). In the case of the UB14 gene in yeast, a series of UBs are attached together.26 In nature, a special de-ubiquitinating enzyme is needed26,28 to extract the UB portion.

The substrate to be degraded with the help of UB must first be identified, and then UB bound specifically to one of the lysine amino acids. In the words of one reviewer,
‘Ubiquitin (Ub) is a conserved 76-amino-acid protein attached posttranslationally to substrate proteins. This conjugation occurs through an isopeptide bond between the C-terminal carboxylate of Ub and the epsilon-NH2 of a lysine side chain in the target protein. Conjugation is achieved by the sequential action of an E1 activating enzyme, E2 conjugating enzymes, and E3 ligases. The removal of Ub from substrates is carried out by deubiquitinating enzymes.’29
The abbreviations E1, E2 and E3 are used commonly in the UB literature, and many variants exist in the same organism, each with special functions. These are very complex enzymes and are much larger than UB. Portions of the folded UB must interact precisely in three dimensions with the three classes of enzymes. The whole scheme is only useful once carefully regulated. Indiscriminately destroying proteins that contain the amino acid lysine would quickly destroy the cell.
UB is involved in a large number of cellular processes. Cell cycle regulation, growth control, development and the stress response require various regulatory proteins to be degraded. This is accomplished by attaching UB, with the help of the three classes of enzymes mentioned, to these substrates.30 UB-conjugated proteins are thus targeted and then degraded in a complex machine called a proteasome.30 Ubiquitination also targets cell-surface proteins to help ingest them for subsequent degradation in lysosomes.31
The proteasome is composed of two sub-complexes: a 20S core particle (CP) that performs the catalytic activity and a regulatory 19S regulatory particle (RP). The 20S CP is a barrel-shaped structure composed of four stacked rings, two identical outer α-rings and two identical inner β-rings. The eukaryotic α- and β -rings are composed each of seven distinct subunits.26
This overview covers only some of the signalling functions UB is involved in, and demonstrates that postulating its origin by evolutionary processes would require also considering where all the other components it is used with came from, and in which order.

4. Refined to 1.8 Å, coordinates from the Protein Data Base www.rcsb.org/pdb, entry 1UBQ.pdb. Displayed with Swiss Prot protein viewer. 5. Refined to 1.8 Å, coordinates from the Protein Data Basewww.rcsb.org/pdb, entry 1UBQ.pdb. Displayed with RasTop protein viewer. Backbone structure emphasized. 6. Another view, based on the same data as Figure 4. The PES residues and location of lysines are shown. Displayed with Swiss Prot protein viewer.
Interpretation of the Ubiquitin data
All scientific theories are subject to revision, although this is often resisted vehemently.32 We must not overlook the fact that errors in reported sequences are an unfortunate reality in protein and gene databases. In many cases data is available from only a single source. Most organisms have multiple copies of UB, and one mutated version might not be functional. UB from parasites, which may rely heavily on their host’s genetic equipment, might not work in free-living organisms. Viruses were found to sometimes contain UB-resembling fused proteins. Since the rest of the enzymes needed to tag substrates and to create a proteasome were missing, it is not clear if these sequences actually serve any function. UB found in viruses were excluded from this study.
Some design features become apparent when we examine the folded structure of UB (Fig. 4). The distinctive end amino acid triplet (-RGG) is exposed on the outer surface of the protein. The three amines of R (Arginine) render this residue strongly hydrophilic. And the G (Glycine) side chain is the smallest possible for amino acids, a simple hydrogen. The combined effect is to expose the terminal COOH group involved in isopeptide bonding in the aqueous media of the cell, unhindered and distant from the hydrophobic protein core (Fig. 5). The NH2 of Lys-48, involved in multiple ubiquitination links, is accessible to the COOH group. M-1 (Methionine) is protected by the central hydrophobic core.
The PES pattern at positions 19–24–57, can be viewed (Fig. 6) from another perspective. Proline-19 and Serine-57 are shown to be reasonably close together when UB is folded. Perhaps both interact with one of the enzymes. Proline is distinctive among amino acids in that peptide bonds form a sharp kink in the protein backbone (since no free hydrogen remains attached to the amide). It is possible that an E1 or E2 enzyme is tailored to conform to this feature for most animals but not other kinds of organisms.
It is remarkable that the very small differences revealed by the diagnostic patterns of animal, plant and fungus could be so important. The associated enzymes are presumably very precisely engineered to match.
An evolutionary interpretation would be that once near-perfection has been attained natural selection limits the amount of variability which will result afterwards. But if the selectivity differences between near-perfect and close variant are indeed so dramatically different, how would the preceding organisms have survived without UB?
One creationist interpretation would be that God designed alternate UB variants, and their associated enzymes, for different taxa. This would modify how fast various proteins are targeted for degration (the ‘Nend Rules’), optimal for the needs of those kinds of organism. Mutations over time would modify a few less critical parts of UB.
Various lysines (K) on UB serve different purposes. It is noteworthy that the end-carboxyl group of UB can distinguish, with the help of various enzymes, among the seven K residues present to form poly-ubiquitin chains using primarily the one at position 48, but also the one at position 29 to degrade other proteins. A linkage with K63 serves as a proteolysis-independent signal for several processes. It is also noteworthy, that the NH2 of other side-chain amines (asparagine, glutamine and arginine) of other proteins are recognized by the enzymes as incorrect targets. In addition, a single amino acid at position 57 distinguishes plants from fungi. The serine (S) side chain of fungi has a chemical functional group, H2C-OH, which is polar and can form weak H-bonds, but nevertheless is very close in size to the side-chain of the plants’ alanine (A), CH3. This implies the enzyme variants are precisely tuned so that they can interact upon such a small differences.
Substituting proline by alanine, as found in bees, one cockroach and some worms, leads to a small but observable difference in a portion of the folded UB.
The oldest cockroach fossil is claimed to be about 300 million years old, and the oldest bee 80 million years old. The best evolutionist interpretation would involve independent and coincidental mutations and not a common ancestor for cockroaches, bees and some worms.
The classical YEC model predicts a severe, worldwide, bottleneck in cockroach and bee populations caused by turbulent conditions during and following a universal Flood. This period could have lasted hundreds of years.33 A few founder members possessing the minority AES pattern might have survived the Flood, and their lineages survived through genetic drift in the small populations. As an alternative explanation, the latest thinking, based on the RATE project34 results, is that a short period of high radioactivity accompanied the Flood event, and may have caused a large number of mutations throughout the biosphere. P → A may have occurred during this time. Some of these could easily fix rapidly in the very small effective populations.
Bees are unusual in many ways, such as a unique manner by which sex is determined.35 Shortly after the Flood, in which few bee colonies would have existed, one of those carrying a mutant UB may have fixed in the population. In-breeding among direct descendents of the same queen and the extremely small number of reproductive members would make it easy for a mutation to fix.
Notice that worms are hermaphrodites, facilitating fixation of mutations.
The extra amino acids at critical locations present in Cercozoa preclude an evolutionary scenario: animal → Cercozoa → plants (P E S → S E A → S D A ).
What we lack at this time is an extensive dataset with a much wider range of organisms, especially those closely related (such as different species of cockroaches). We also lack a wide range of sequences from the same species, to see how much variability from the species-specific consensus sequence exists. Until more data becomes available, one cannot be sure that the animal AES pattern is not a deliberately designed feature.
UB and the evolutionary time scale

Figure 7. Diagnostic ubiquitin pattern for animal, fungus and plant life-forms show essentially no variability within these three lineages. There is no evidence ubiquitin arose from a single gene sequence. (Evolutionary ages assumed). (Click on picture to see a larger version)
The very small amount of variability in functional UB sequences is problematic for the evolutionary viewpoint, and has been exacerbated by the finding of multiple families of these proteins (see Fig. 7).
Current evolutionary thinking is that the first eukaryote cell lived about 2.7 thousand million years ago.36 Prokaryotes don’t possess anything resembling UB, and most evolutionary biologists still believe eukaryotes arose from them. Based on the distinguishing UB patterns from my dataset, plants (SDA) and fungi (SDS) are more similar to each other than to animals (PES). Using other genes and assuming that greater sequence difference implies greater time divergence from a common ancestor leads to the opposite conclusion, that animals are more closely related to fungi (anima-fungi common ancestor: 1513 Ma) than to plants (animal-plant common ancestor: 1609 Ma).36 In other words, 1.609 thousand million years ago UB must have already been present. One still needs a common ancestor with the UB version used by Cercozoa and Foraminifera, and presumably this first UB didn’t immediately pop into existence. This is illustrated in the top portion of Fig. 7 whereby over 1.61 thousand million years ago a theoretical common ancestor would possess the same sequence for some period of time. Since evolutionists constantly change the date they wish to use for a common ancestor for Cercozoa and Foraminifera, I cannot extrapolate to a common ancestor with the animal-fungus-plant lineages. According to analysis based on small-subunit rDNA sequences,37 Foraminifera were claimed to branch near the base of the eukaryotic tree, hundreds of million years before the animal-plant split. Note that Foraminifera do possess fully functional UB. Subsequent authors have proposed a less ancient origin.
An evolutionary interpretation of the data leads to the conclusion that UB must have been present in the first eukaryote or showed up shortly afterwards. In other words, four lineages have remained virtually unchanged for about 1.5 thousand million years. But for the creation of the very first functional UB and the many other necessary proteins far less time would have been available.
In a later paper I will introduce the sequence data available for UBLs, which together with our present analysis of UB renders the evolutionary interpretive framework for their chance origin very problematic.
Conclusions
We see that instead of 4 thousand million years to produce a DNA sequence to code for ubiquitin, using the evolutionary model in an internally consistent manner requires it to have been already present in the first eukaryote or to have arisen unrealistically quickly afterwards. This seems rather miraculous, given the complexity of the bio-chemical processes UB is involved in, and the need for E1, E2, E3, de-ubiquitination enzyme and the proteasome concurrently. These enzymes are precisely tuned to the folded UB structure, as reflected by the intolerance of UB to mutations. These enzymes have no other known function and do not interact with the UBLs, the putative precursors of UBs. Furthermore, the ‘Nend Rules’,26 which determine the half-life of proteins, would need to be coordinated across unrelated proteins for the good of the organism as a whole. Until perfected, the UB-based degradation would be very deleterious.
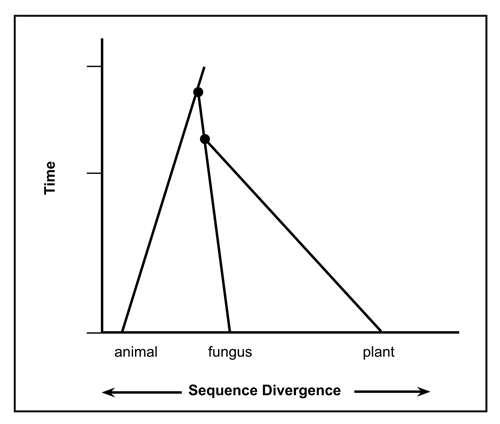 Figure 8. Phylogenic evolutionary expectations assuming the most parsimonious divergence of the diagnostic ubiquitin pattern for animal, fungus and plant life-forms. |
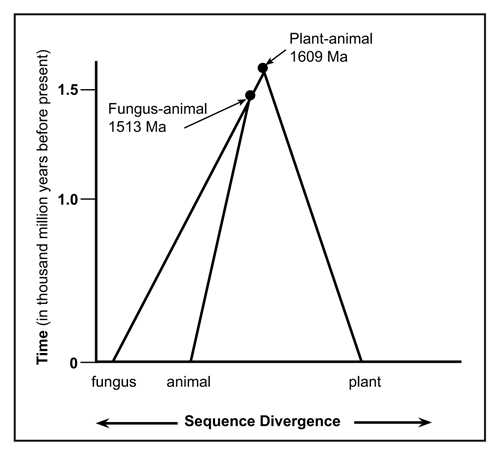 Figure 9. Phylogenic relationship between animal, plant and fungus life-forms according to current evolutionist theory based on several genes. Data from ref. 37. (Click on picture to see a larger version) |
Given the large number of key regulatory processes UB is involved in, it is indispensable for this cell type. Prokaryotes have an unrelated scheme to degrade regulatory proteins.
Analysis of the available data shows that there are at least four families of ubiquitin. From the sequence variations one could reasonably conclude that from the beginning of their existence, animals, fungi and plants always possessed the appropriate UB pattern (Fig. 7). The UB sequence evidence does not support the claim that these diverged from a common ancestor. Analysis using UB, makes fungi-animalia seem less similar (PES-SDS) than fungi-plantae (SDS-SDA) (Fig. 8), whereas interpretation of sequence data from other genes with evolutionary spectacles implies the opposite (Fig. 9).
The insignificant range of sequence variability found within each family of ubiquitin indicates that claiming an origin through Darwinian processes is unreasonable. The chances of coding for a minimally first version natural selection could operate on, ca. 4X10-83, are infinitesimally small.
Perhaps there are actually far more acceptable variants of UB than we estimated, and possibly the mutational rates were much greater in the past, such as 10-6 per base pair. It becomes obvious, however, that with these assumptions we still don’t make even a small dent towards overcoming the improbability of being able to code for an initial UB by chance. And this highlights a second difficulty evolutionary theory faces: why then is there so little variability after more than a thousand million years of mutations across several independent lineages? During this time, on average every base pair of UB would have mutated more than 1041 times,38 generating widespread polymorphisms and fixed alternatives by now.
In Table 27 the actual amount of variability should be considered, keeping in mind to which of the four identified families of UB the organism belongs to. Variability in residue positions 19, 24 and 57 can pretty much be ignored, since they classify the UB families. The remaining variability can be to a first approximation distributed among three groups (animal, fungus and plant).
Evolutionary theory would imply that these lineages have remained dramatically invariant on average for about 1.5 thousand million years. A creationist would argue the data demonstrate very little sequence variability and ubiquitin is involved in complex processes requiring precise tuning of many other proteins concurrently before it could work—a wonderful example of irreducible complexity.
Appendix 1
Probability calculations using information theory
Let us assume that our dataset is representative of the amount of variability permitted by a functional ubiquitin. The Shannon Information Theory calculations used below have been explained in this journal already.9 The method decreases the number of possible random polypeptides 76 amino acids long, arguing that many of these belong to a very low probability set.9 This increases the probability of finding an initial one by chance, which natural selection could then fine-tune.
Define pj as the probability an acceptable amino acid is found at a residue position ‘l’ on the UB protein. Using the genetic code, we weight according to synonymous codes Σpj over all amino acids tolerated at a given residue position. For each site we calculate:
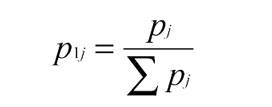
(1)
from which the entropy at each site l is calculated as:

(2)
For my dataset, an entropy of 40.94 bits for the 76 residues is calculated.
Using an approximation of pij based on the distribution of the twenty natural amino acids, leads to an estimate of the entropy of DNA over all genomes9,17 of 4.139 bits, based on (3):

(3)
Then the effective total number of DNA sequences (excluding those in the low probability set) is estimated to be:

(4)
The effective number of functional UB sequences we calculate as:

(5)
and the proportion of functional UB sequences compared to random polypeptide chains 76 residues long is the ratio of (5) / (4): 4.3X10-83.
References
- www.ncbi.nlm.nih.gov/BLAST/; protein-protein BLAST (blastp) tool used. Return to text.
- Free ActivePerl software can be downloaded from: www.activestate.com/Products/Download/Download.plex?id=ActivePerl, 20 September 2005. Return to text.
- https://dl0.creation.com/articles/p043/c04346/appendix_a_b_jun_28_2005.txt, (Appendix A and Appendix B). Return to text.
- Free ClustalX software can be downloaded from: www-igbmc.u-strasbg.fr/BioInfo/ClustalX/Top.html, 20 September 2005. An example of multiple sequence alignment using some ubiquitin sequences. Return to text.
- https://dl0.creation.com/articles/p043/c04346/ubfigure01small.png. Return to text.
- Online data for ubiquitin sequences is provided at: creation.com/images/journal_of_creation/vol19/ table_ubiqutin_full_set_june_25_2005.xls. Return to text.
- Online table with summary of variability of ubiquitin sequences: creation.com/images/journal_of_creation/vol19/ubtableb.htm. Return to text.
- Online Excel spreadsheet with all variability of ubiquitin sequences creation.com/images/journal_of_creation/vol19/ table_ubiquitin_variable_residues_june_25_2005.xls. Return to text.
- Truman, R. and Heisig, M., Protein families: chance or design? TJ 15 (3):115–127, 2001. Return to text.
- Number of alternative amino acids present at each residue position of Table 3: 3x3x2x3x5x4x4x2x4x5x6x3x2x3x3 = 167961600, which assumes context independence for data reported as of August 2004. Note that only 55 alternatives in total were actually identified in Table 3. The true number of functional alternatives according to Table 3 is probably much lower than about 168 million. For example, of the 158 organisms reported in the dataset, many are identical. Exhaustive examination of functional ubiquitin alternatives (ref. 1) during May of 2005 for the same organism always showed less than 10 alternatives reported so far, implying < 1582x10 variants would be predicted based on existing data as shown in Table 3. Assuming context independence has dramatic consequences, which increases with the number of residue positions displaying variability. Return to text.
- Ref. 5, see organisms with id 39 and id 123. Return to text.
- Ref. 5, see organisms with id 39, 152 and 153. Return to text.
- Ref. 5, see organisms with id 147 and id 148. Return to text.
- Ref. 5, see organisms with id 127 and id 130. Return to text.
- Archibald, J.M., Longet, D., Pawlowski, J. and Keeling, P.J., A novel polyubiquitin structure in Cercozoa and Foraminifera: evidence for a new eukaryotic supergroup, Mol. Biol. Evol. 20 (1):62–66, 2003. Return to text.
- Scherer, S. and Loewe, L., Evolution als Schöpfung? in: Weingartner, P. (Ed.), Ein Streitgespräch zwischen Philosophen, Theologen und Naturwissenschaftlern , Verlag W. Kohlhammer, Stuttgart; Berlin; Köln: Köhlhammer, pp. 160–186, 2001. Return to text.
- Yockey, H.P., Information Theory and Molecular Biology , Cambridge University Press, Cambridge, p. 250, 1992. Return to text.
- Fersht, A.R., DNA replication fidelity, Proceedings of the Royal Society ( London ) B 212 :351–379, 1981. Return to text.
- Drake, J.W., Charlesworth, B., Charlesworth, D. and Crow, J.F., Rates of spontaneous mutation, Genetics 148 , 1667, 1998. Return to text.
- Grosse, F., Krauss, G., Knill-Jones, J.W. and Fersht, A.R., Replication of fX174 DNA by calf thymus DNA polymerase a: measurement of error rates at the amber-16 codon, Advances in Experimental Medicine and Biology 179 :535–540, 1984. Return to text.
- Spetner, L., Not by Chance! Shattering the Modern Theory of Evolution , The Judaica Press, Inc., Chapter 4, 1998. Return to text.
- Wagner, A., Energy constraints on the evolution of gene expression, Mol. Biol. Evol. 22 (6):1365–1374, 2005. Return to text.
- As an alternative constraint-based approach to evolutionary scenarios, ReMine has clarified Haldane’s dilemma. Reproductive excess is needed to cover all ways organisms can perish (ref. 33) and more offspring are needed if a favoured mutant is to out-populate the original type. The number of offspring necessary to accommodate evolutionary scenarios becomes unrealistically high. Return to text.
- Generally, not more than one new amino acid would mutate per gene per generation. An exact analysis would require taking residue context into account. Multiple changes producing compensatory effects cannot be tested effectively by evolutionary processes, which generally only changes a single amino acid at a time. In other words, a very limited subset of possible solutions can be tested by trial and error.
Suppose five specific amino acids need to be modified to generate a protein with a new function. Out of 20 possible amino acids at each of the five positions, generally several are acceptable. The probability of a single mutation occurring, which provides one of the acceptable amino acids, would on average be about 10-10 per organism. These odds must be overcome 5 times. What are the odds of such a transformation occurring? Assuming all organisms which ever could have lived were involved in the effort produces a probability of 1046 x 10-50 = 0.0001.
Of course, additional undesirable mutations are also likely to arise during this time, creating additional hurdles. Return to text. - Unpublished results, paper in preparation. Return to text.
- Glickman, M.H. and Ciechanover, A., The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction, Physiol Rev. 82 :373–428, 2002. Return to text.
- Krebber, H., Wostmann, C. and Bakker-Grunwald, T., Evidence for the existence of a single ubiquitin gene in Giardia lambia , FEBS Letters 343 :234–236, 1994. Return to text.
- Varshavsky, A., The N-end rule and regulation of apoptosis, Nature Cell Biology 5 :373–376, 2003. Return to text.
- Hemelaar, J., Borodovsky, A., Kessler, B.M., Reverter, D., Cook, J., Kolli, N., Gan-Erdene, T., Wilkinson, K.D., Gill, G., Lima, C.D., Ploegh, H.L. and Ovaa H., Specific and covalent targeting of conjugating and deconjugating enzymes of ubiquitin-like proteins, Molecular and Cellular Biology 24 (1):84–95, 2004. Return to text.
- Wilkinson, K.D., Ubiquitin-dependent signalling: the role of ubiquitination in the response of cells to their environment, J. Nutr. 129 :1933–1936, 1999. Return to text.
- Hershko, A. and Ciechanover, A., The ubiquitin system, Annu. Rev. Biochem. 67 :425–479, 1998. Return to text.
- Kuhn, T., The Structure of Scientific Revolutions , 2 nd ed. enl., Chicago University Press, Chicago, 1970. See also: Ratsch, D., The Philosophy of Science: The Natural Sciences in Chris tian Perspective , IVP, Downers Gr., IL, 1986. Return to text.
- Scheven, J., Mega-Sukzessionen und Klima im Tertiär. Katastrophen zwischen Sintflut und Eiszeit , Hänssler, Neuhause-Stuttgart , Germany, 1988. Return to text.
- Vardiman, L., Snelling, A.A. and Chaffin, E.F., Radioisotopes and the Age of the Earth , Institute for Creation Research, El Cajon , CA , 2000. Return to text.
- news.nationalgeographic.com/news/2003/09/0908_030908_beegene.html, 19 September 2005. ‘Why do unfertilized bee eggs become male, while fertilized eggs produce female worker or queen offspring? According to new research, the answer lies in a unique form of genetic sexual determination. Female bees carry two slightly different copies of a sex-determination gene: one from their mother and the other from their father, which may work together to trigger female development. Unfertilized male bee eggs, however, have only a single copy of the gene from their mother, which causes their development to proceed along the male route. This is a totally different genetic system to that discovered in other animals [humans, mice, and some worms and flies for example]&rsquo. Return to text.
- Hedges, S.B., Blair, J.E., Venturi, M.L. and Shoe, J.L, A molecular timescale of eukaryote evolution and the rise of complex multicellular life, BMC Evolutionary Biology 4 :1–9, 2004; www.biomedcentral.com/1471-2148/4/2#B1. Return to text.
- Pawlowski, J., Bolivar, I. , Guiared-Maffia, J. and Gouy, M., Phylogenetic position of the Foraminifera inferred from LSU rRNA gene sequences, Mol. Biol. Evol. 11:929–938, 1994. Return to text.
- (76 amino acids)(3 base pairs/amino acid)(10-6 mutations/base pair x organism)(0.25 x 1046 organisms) = 5.7 x 1041 mutations on average for each base pair of UB. Return to text.
Readers’ comments
Comments are automatically closed 14 days after publication.