

Journal of Creation 25(2):106–110, August 2011
Browse our latest digital issue Subscribe
The chromosome 2 fusion model of human evolution—part 1: re-evaluating the evidence
One of the leading molecular arguments for human evolution from a shared common ancestor with apes, particularly chimpanzees, is the ‘chromosome 2 fusion model’. This scenario involves the claim that the fusion of two small chimpanzee-like chromosomes (2A and 2B) formed one stable chimera chromosome in humans, leading to the difference in diploid chromosome numbers between humans and great apes. A majority of the data for the fusion model is based on DNA hybridization and chromosomal staining experiments conducted prior to the sequencing of the human and chimpanzee genomes. In the present paper, we present a new analysis of the scientific literature, and in a companion paper (part 2 in this issue) a re-analysis of the available DNA sequence data that calls into question the validity of the fusion model.

One of the most popularized molecular arguments for human-primate evolution is the hypothetical prehistoric head-to-head fusion of two primate chromosomes (corresponding to 2A and 2B in chimpanzee) to form human chromosome number 2. Much of the research supporting this hypothetical model is based on indirect evidence derived from DNA hybridization and chromosomal staining techniques. These techniques provide only approximate estimates of sequence similarity, with hybridization-based analyses being more accurate than the analysis of stained chromosomal bands. This type of initial evidence, along with some targeted DNA sequencing of small genomic regions in human, seemed to indicate support for the fusion model.1,2
While the chromosome 2 fusion model is routinely touted as dogma, very little new genomic data, although readily available for analysis, has been presented as evidence. In addition, several science authors have recently published books for the general public popularizing this hypothetical model as one of the supposedly strongest arguments for human evolution from a shared common ancestor with apes, particularly chimpanzees.3,4
Popular reviews on this subject often include a simplified drawing depicting how the putative fusion of two small acrocentric5 ape-like precursor chromosomes could have fused end-to-end to form the larger human chromosome 2, as shown in figure 1. In support of this hypothetical model of chromosome fusion, it is claimed that human chromosome 2 contains two key features that support the model. The first feature purportedly depicts the fusion event and contains genomic sequences representing a head-to-head fusion of telomeres, the highly specific endcap DNA repeat motifs (TTAGGG)n located at the termini of linear mammalian chromosomes.6
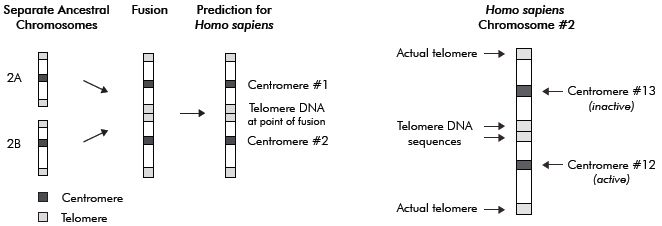
The second key site purportedly represents a cryptic, non-functional centromere that was silenced following the fusion event (because a single functional centromere is required for chromosome stability and function). According to these claims, this fusion event accounts for the fact that humans have only 46 (2N) chromosomes and the great apes 48 (2N). Actually, the diploid genomes of gorilla, chimpanzee and orangutan have 48 but some gibbons have 44, and one Malaysian ape has 50.7 The fusion model scenario involves a hominid evolved from a shared common ancestor with a diploid genome of 48 chromosomes, and, in some early human ancestor, two chromosomes fused, reducing the diploid chromosome complement to 46.
Examining the existing genomic evidence for fusion
Of the two genomic features that are claimed to support the fusion model, the primary evidence used is the presence of a reputed fusion site. This site is located in a pericentric region (meaning it is close to the present functional centromere) on the long arm of human chromosome 2. The DNA sequence at this location is supposed evidence of a head-to-head telomeric fusion of two acrocentric chromosomes.
In his recently published book, the Nature article Miller cites as proof for the fusion states only that “Human chromosome two is unique to the human lineage in being the product of a head-to-head fusion of two intermediate-sized ancestral chromosomes” and provides no evidence for this conclusion. However, Fairbanks offers more detail and claims that there appears to exist a fusion site involving a set of 158 telomere sequences, and, of the 158 repeats, he notes only 44 sets can be manipulated to achieve perfect telomere consensus sequences. Another example of the same claim from a book popularizing human evolution is as follows:
“The DNA sequences in the human chromosome are exactly as expected from this scenario. Telomeres consist of many repeats of the nucleotide sequence TTAGGG, and at the fusion point of the human chromosome, where the two telomeres fused, this sequence is found ‘head to head’. The functional centromere in chromosome 2 lines up with the chimpanzee chromosome 2p13 chromosomal centromere. The remains of the redundant centromere from one of the ancestral ape chromosomes can also be found.”3
As we will document, these popular claims of resounding evidence for a telomere fusion producing human chromosome 2 are, for the most part, unsupported by the scientific literature and actual DNA sequence information (see companion paper). However, we will first briefly clarify the structure and nature of telomeres as to what would be expected if such a fusion event occurred. In so doing, we will take into account the accepted evolutionary presuppositions and timelines related to such an event.
Telomeres are typically found at the ends of linear eukaryotic chromosomes and confer stability by preventing fusion via a ‘capping’ function. The telomere region involves a complex and dynamic framework of DNA motif repeats, structural loops, structural and functional RNAs and a wide variety of proteins.6 In a fusion event as described in the human chromosome 2 model, the end result should produce two identifiable telomeres characterized by a specific repeat motif and oriented in a head-to-head configuration. A certain genomic landscape must be present if two chromosomes fused head-to-head as claimed by the current evolutionary model. The consensus 5’ to 3’ telomere motif in humans, chimps, apes, and mammals in general, is (TTAGGG)n and typically occurs in perfect tandem for stretches of DNA from about 10 to 15 kb (10,000 to 15,000 bases) and contains 1,667 to 2,500 telomere repeats at each chromosome end. In a head-to-head fusion of two chromosomes, we would expect at least 5,000 bases of (TTAGGG)n repeats in tandem, albeit in a slightly degenerate state, given a supposed ~1 to 5 million years of evolution since the fusion event occurred. At the point of fusion, we would also expect the orientation of the plus-strand repeat to change to the reverse complement (CCCTAA)n, which should also occur in near-perfect tandem for approximately 5,000 or more bases.
In reality, the putative fusion site is but a vague shadow of what should be present given the model in question. One of the major problems with the fusion model is that, within the 10 to 30 kb window of DNA sequence surrounding the hypothetical fusion site, a glaring paucity of telomeric repeats exist that appear mostly as independent monomers, not tandem repeats. Based on the predicted model, thousands of intact motifs in tandem should exist. For the TTAGGG repeat to the left of the fusion site, less than 35 motifs exist, a normal human telomere would typically have 1667 to 2500.6 For the CCCTAA reverse complement sequence, to the right of the fusion site, less than 150 telomere motifs can be found. Another problem with these two motifs, that we document in our companion research paper, is that their occurrences are found scattered throughout both sides of the fusion site where they would not be expected. In other words, both the forward and reverse complement of the telomere motif populate both sides of the fusion site.
Besides the extreme paucity of telomeric repeats, their largely monomeric condition and their ubiquitous presence on both sides of the purported fusion site, there is very little to indicate that they once formed 10- to 15-kb stretches of perfect tandem 6-base repeats. If a fusion occurred, the alleged sequence no longer resembles telomeric repeats, a problem explained away by fusion supporters by claiming the telomeric repeat area is incredibly degenerate. Nor is the location of the alleged telomeric repeats in the region where it should be, judging by our analysis of the chromosome it is claimed to be fused to (covered in part II of the study).
The only evolutionary research group to seriously analyze the actual fusion site DNA sequence data in detail were confounded by the results which showed a lack of evidence for fusion—a genomic condition for this region which they termed ‘degenerate’.8 In attempting to correlate rates of evolutionary change with the extreme degeneracy observed in the putative fusion region, they claimed that the “head-to-head arrays of repeats at the fusion site have degenerated significantly from the near perfect arrays of (TTAGGG)n found at telomeres.” They also stated, “if the fusion occurred within the telomeric repeat arrays less than ~6 Ma, why are the arrays at the fusion site so degenerate?” The actual data indicates that perhaps the only thing that is degenerate is the evolutionary dogma surrounding the fusion model.
Because of the ‘degenerate’ nature of the DNA sequence data in this region, a variety of creative and manipulative approaches have been used to make the data look more telomere-like than it actually is. For example, Fairbanks claims that 44 out of 158 repeats match (28%) and that the rest of the sequences are ‘close’.3 The problem is, to obtain even this low match level, the consensus reading frame is entirely ignored and ambiguous matches are contrived by assuming many insertion and deletion mutations of varying sizes. In addition, Fairbanks’ data include several additional perfect motifs immediately surrounding the fusion site that do not actually appear in the current GenBank accessions for this region.3 Unfortunately, Fairbanks did not cite the accession number(s) for his fusion site sequence printed in his text. When the reading frame is corrected at various motifs near the fusion site, hardly any telomere sequences can be obtained. Fairbanks assumes that major differences between a perfect telomere and the existing sequence are the result of the accumulation of numerous insertions, deletions and other mutations, a post-hoc explanation that lacks strong DNA evidence.
Another problem for the fusion theory is the presence of a wide variety of genes throughout the fusion region. At present, no known protein coding genes have been found in the 10 to 15 kb tandem 6-base (TTAGGG) repeat terminal region of human telomeres.9 In an analysis of a 614 kb area encompassing the postulated chromosome fusion site, Fan et al. found evidence of “at least 24 potentially functional genes and 16 pseudogenes”.10 In the 30-kb region directly encompassing the fusion site, which should definitely be devoid of any genes, there exists two actively transcribed genes, each in a flanking position in regard to the fusion site (one on each side). There are also at least two other genes in the immediate vicinity of the fusion site thought to be inactive due to frame shift mutations. However, research related to the human ENCODE (Encyclopedia of DNA Elements) project has shown that many genes thought to be inactive (pseudogenes) are actually functional due to a variety of newly discovered regulatory mechanisms.11
If the telomere motifs that populate internal areas of chromosomes serve some important, yet unknown function, the chromosome fusion model actually impedes research aimed at determining possible function in these regions. This type of reasoning is not without precedent. For example, the widely held concept of the genome consisting of mostly ‘junk DNA’ has now been discredited.11,12
Assuming that two telomeres exist in a head-to-head fusion produces another major problem, namely that telomeres are designed to prevent fusion. Broken chromosomes at any location immediately invoke the cell’s double-stranded DNA repair machinery where the aberrant fusion of fragments actually triggers cell fault tolerance mechanisms.6 In the case of an aberrant fusion, a senescence response or programmed cell death (apoptosis) cascade is normally triggered, effectively eliminating the damaged cell from the system.
A cell with telomeres that have progressively shortened over time and reached a threshold length will also activate the double-stranded DNA repair machinery; inducing cell senescence and/or death. When in certain types of germline cells, telomerase adds telomere repeats to shortened telomeres, chromosomes are ‘healed’ and can again become stable. The telomeres cap the ends of linear chromosomes and effectively prevent fusion or trigger cell elimination if the telomere is shortened to a certain point, damaged, or aberrantly fused.6 According to the fusion model, this protective process was somehow bypassed in early humans.
Examining the evidence for a cryptic centromere
Yet another major problem with the fusion model is the lack of evidence for a cryptic second centromere site. Immediately following the supposed head-to-head telomere fusion, there would have existed two centromeres in the newly-formed chimeric chromosome, one from each of the two fused chromosomes. This type of event had to occur in a cell lineage of the germ-line to be heritable, and one of the centromeres would have had to be immediately eliminated or at least functionally silenced for cell division to progress normally. Evolutionists explain the lack of a clearly distinguishable non-functional secondary centromere by arguing that two centromeres would result in major instability when chromosomes pair up during cell division and consequently would be rapidly selected against. According to the evolutionary model, selection would continue until the second centromere was completely non-functional.
However, the evidence for a second remnant centromere at any stage of sequence degeneracy is negligible. As Fairbanks noted, “Fusion at the telomeres should have left two centromeres in the ancient fused chromosome, but there is only one now.”3 He then evaluated the “evidence that a centromere was once present at a second site”. The supposed evidence includes the finding that “every human and great-ape chromosome centromere contains a highly variable DNA sequence that is repeated over and over, a 171 base-pair sequence called the Alphoid sequence.”3 Fairbanks adds that scientists have “searched for Alphoid sequences in human chromosomes and found them at every centromere, as expected. They also found Alphoid sequences at the site in human chromosome 2 where the remnants of this second centromere should be. These remnants are evidence of a now-defunct centromere.”3
The main problem with Fairbank’s claim is that alpha-satellite DNA or alphoid DNA, although found in centromeric areas, is not unique to centromeres and is also highly variable. Because highly variable alphoid DNA is also commonly found in non-centromeric regions of human chromosomes, their presence does not indicate the remnants of a degenerate centromere.
Based on the reasoning of Fairbanks and others promoting the human chromosome 2 fusion model, one could conclude that human chromosomes contain literally hundreds of degenerate centromeres. As a result, locating a candidate alphoid region to erroneously support the presence of a degenerate centromere on chromosome 2 would not be unexpected or difficult to do, and does not support a cryptic centromere claim. In the companion research paper supporting this review, we show that the alphoid sequence in question does not align closely with known functional centromeric human DNA.
Despite the variation, there is enough sequence similarity for fluorescently labeled alphoid probes to hybridize to most classes of alphoid sequence in stretched chromosomal fibers. This explains many of the early chromosome fusion reports which relied on this technology (as shown in part II alphoid sequences are found throughout large sections of the chromosome and are not by themselves evidence of a centromere.).1,2 Another problem is that, although research has been done on some primates, no systematic study of centromeres exists to determine how common alphoid DNA is in mammals.13
Multiple reports of alphoid/centromere similarity between humans and apes, involving both hybridization and sequence-based research, find that there is virtually no apparent evolutionary homology, except for moderate similarity on the X-chromosome centromere.13 Baldini et al. found that the “highest sequence similarity between human and great ape alphoid sequences is 91%, much lower than the expected similarity for selectively neutral sequences.”13 Alphoid regions, in contrast to many classes of DNA sequences, are not well-conserved among mammalian taxa and even show high levels of diversity between chromosomes in the same genome.14
Cytogenetic anomalies argue against fusion
Other problems with the fusion theory include the fact that standard cytogenetic techniques, such as C-banding, have detected significantly less heterochromatic centromeric DNA on the long arm of human chromosome 2 than predicted by the fusion model. Evolutionists claim this is because the “bulk of the centromeric repetitive DNA has been lost”.13 Conversely, it is more likely that the so-called cryptic centromeric DNA never existed.
Not only does the DNA sequence at the putative cryptic centromere site argue against fusion, but a comparison of the chimp and human chromosomes reveals that the centromere in human chromosome 2 is in a very different location than predicted by a fusion event as shown in part two of this study. This necessitates an implausible series of events, including the loss of both chimp centromeres when chromosomes 2A and 2B fused, and the rapid evolution of a new centromere to provide functionality to human chromosome 2.
Mutations of the magnitude needed to support a fusion event pose serious cytogenetic problems both for the organism during regular somatic cell growth related to mitosis and during the meiotic events occurring in the germ-line tissues. Proper alignment requires the near-identical structure of each pair so that each chromosome aligns only with its sister chromosome. Chromosomal fusion is one major common cause of infertility. If meiosis does occur despite the aberration, the embryo produced from fertilization of these gametes typically self-aborts.15
Do fusions lead to new species?
Evolutionary scientists believe an ape-like ancestor evolved into a new species, called Homo sapiens, along with a major genomic fusion event. While the order of genes and their spatial relationship in the nucleus can affect gene expression, no new information or genes are added by fusing two existing chromosomes, because only the gene packaging is altered. However, the information content of the genome can still be strongly affected. Chromosomal fusion has been identified in a variety of animal taxa, such as ruminants (sheep, goats and cattle) that were phenotypically similar compared to normal animals in their genera although reproductive isolation did occur.16 Evolutionists postulate that such an event may have contributed to a reproductive barrier in early evolving humans who, although they may have had a new karyotype, were still closely related to apes.
Actually, the fusion theory creates problems for Darwinists due to the fact that a complete absence of humans with 48 chromosomes exists. Although very rare, chromosome fusions do occasionally occur in humans but are not easily passed to offspring. If a chromosomal split occurred during human evolution, then two distinct human groups would result. One evolutionary explanation for this problem is that the entire population of 48-chromosome proto-humans became extinct. Altered animal karyotypes that are not detrimental are rare, but produce populations representing both karyotypes.
Recent genomic problems for fusion
A common claim for fusion is that “the DNA sequence of the rest of human chromosome 2 closely matches very precisely the sequences of the two separate chimpanzee chromosomes.”17 This claim is unsupported by a lack of detailed comparative DNA sequence data. It is noteworthy that the chimp rough-draft DNA sequence assembly was largely based on the human genome as a framework for its construction.18 One of the first published attempts at reporting a genome assembly based on a physical framework19 constructed for the chimp genome was the recently reported chimp Y chromosome project.20 The end result was a completely different and unexpected genomic landscape showing extreme DNA sequence dissimilarities (30% or greater difference) between the human and chimp Y chromosomes. Why comparisons have not been reported for the other chromosomes is unknown, considering the technology to provide a chimp genome assembly based on a chimp contig-based physical map has been available since 2006.20,21
Conclusion
The purportedly overwhelming DNA evidence for a fusion event involving two primate chromosomes to form human chromosome 2 does not exist, even without the aid of new analyses. In this report, our review of only the reported data by evolutionary scientists shows that the sequence features encompassing the purported chromosome-2 fusion site are far too ambiguous to infer a fusion event. In addition to a lack of DNA sequence data for a head-to-head chromosomal fusion, there also exists a decided paucity of data to indicate a cryptic centromere. In a companion paper (part 2) to this, we report the results of additional data analyses using a variety of bioinformatic tools and publicly available DNA sequence resources that further refute the hypothetical chromosome fusion model.
References
- Yunis, J.J. and Prakash, O., The origin of man: a chromosomal pictorial legacy, Science 215:1525–1530, 1982. Return to text.
- Ijdo, J.W. et al., Origin of human chromosome 2: an ancestral telomere-telomere fusion, Proc. Natl. Acad. Sci. 88:9051–9055, 1991. Return to text.
- Fairbanks, D.J., Relics of Eden, Quotations and analyses are in reference to Chapter 1, Fusion, Prometheus Books, Amherst, N.Y., pp. 17–30, 2007. Return to text.
- Miller, K.R., Only a Theory: Evolution and the Battle for America’s Soul, Viking, New York, 2008. Return to text.
- Acrocentric chromosome has one arm that is considerably shorter than the other arm. The ‘acro-’ in ‘acrocentric’ refers to the Greek word for ‘peak’. The human genome contains five acrocentric chromosomes: 13, 14, 15, 21 and 22. The naming system of chimpanzee chromosomes ‘2A’ and ‘2B’ was first used by McConkey, E.H., Orthologous numbering of great ape and human chromosomes is essential for comparative genomics, Cytogenet Genome Research 105:157–158, 2004. Prior to that, the chimpanzee chromosomes were numbered according to size, as is the system used for all other species. Return to text.
- Tomkins, J. and Bergman, J., Telomeres: implications for aging and evidence for intelligent design, J. Creation 25(1):86–97, 2011. Return to text.
- Jauch, A. et al., Reconstruction of genomic rearrangements in great apes and gibbons by chromosome painting, PNAS 89:8611–8615, 1992. Return to text.
- Fan, Y. et al., Genomic structure and evolution of the ancestral chromosome fusion site in 2q13-2q14.1 and paralogous regions on other human chromosomes, Genome Research 12:1651–1662, 2002. Return to text.
- Devanshi, J. and Promisel J.P., Telomeric strategies: means to and end, Annu. Rev. Genet. 44:243–69, 2010. Return to text.
- Fan, Y. et al., Gene content and function of the ancestral chromosome fusion site in human chromosome 2q13-2q14.1 and paralogous regions, Genome Research 12:1663–1672, 2002. Return to text.
- The ENCODE Project Consortium, Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project, Nature 447:799–816, 2007. Also see the June 2007 (vol 17) issue of Genome Research. Return to text.
- Wells, J., The Myth of Junk DNA, Discovery Institute Press, Seattle, WA, 2011. Return to text.
- Baldini, A. et al., An alphoid DNA sequence conserved in all human and great ape chromosomes: evidence for ancient centromeric sequences at human chromosomal regions 2q21 and 9q13, Human Genetics 90:577–583, 1993. Return to text.
- Alkan, C., et al., Genome-wide characterization of centromeric satellites from multiple mammalian genomes, Genome Research 21:137–145, 2011. Return to text.
- Alberts, B. et al., The cell cycle; in: Molecular Biology of the Cell, 5th ed., Garland Science, New York, pp. 1053–1114, 2008. Return to text.
- Lightner, J.K., Karyotype variability within the cattle monobaramin, Answers Research J. 1:77–88, 2008. Return to text.
- Alexander, D., Creation or Evolution: Do We Have to Choose? Monarch, Oxford, UK, p. 212, 2008. Return to text.
- The Chimpanzee Sequencing and Analysis Consortium, Initial sequence of the chimpanzee genome and comparison with the human genome, Nature 437:69–87, 2005. Return to text.
- Warren, R.L., Physical map assisted whole-genome shotgun assemblies, Genome Res. 16:768–775, 2010. Return to text.
- Hughes, J.F. et al., Chimpanzee and human Y chromosomes are remarkably divergent in structure and gene content, Nature 463:536–539, 2010. Return to text.
- genome.wustl.edu/genomes/view/pan_troglodytes/. Return to text.


Readers’ comments
Comments are automatically closed 14 days after publication.