

Journal of Creation 33(1):3–4, April 2019
Browse our latest digital issue Subscribe

More evidence for the reality of genetic entropy—update
Several years after the publication of “A new look at an old virus: patterns of mutation accumulation in the human H1N1 influenza virus since 1918”1 and the follow-up publication in Journal of Creation,2 some detractors are arguing that the analysis was invalid. Specifically, they are claiming that the human H1N1 influenza virus is still making people sick, and thus the claim that it went ‘extinct’ in 2009 was wrong. This was an intriguing thought. Maybe the human H1N1 virus had still been circulating but was missed by major epidemiological laboratories worldwide? To get at the root of this, I re-did the original analysis and extended it to include all H1N1 viral genomes reported to date.
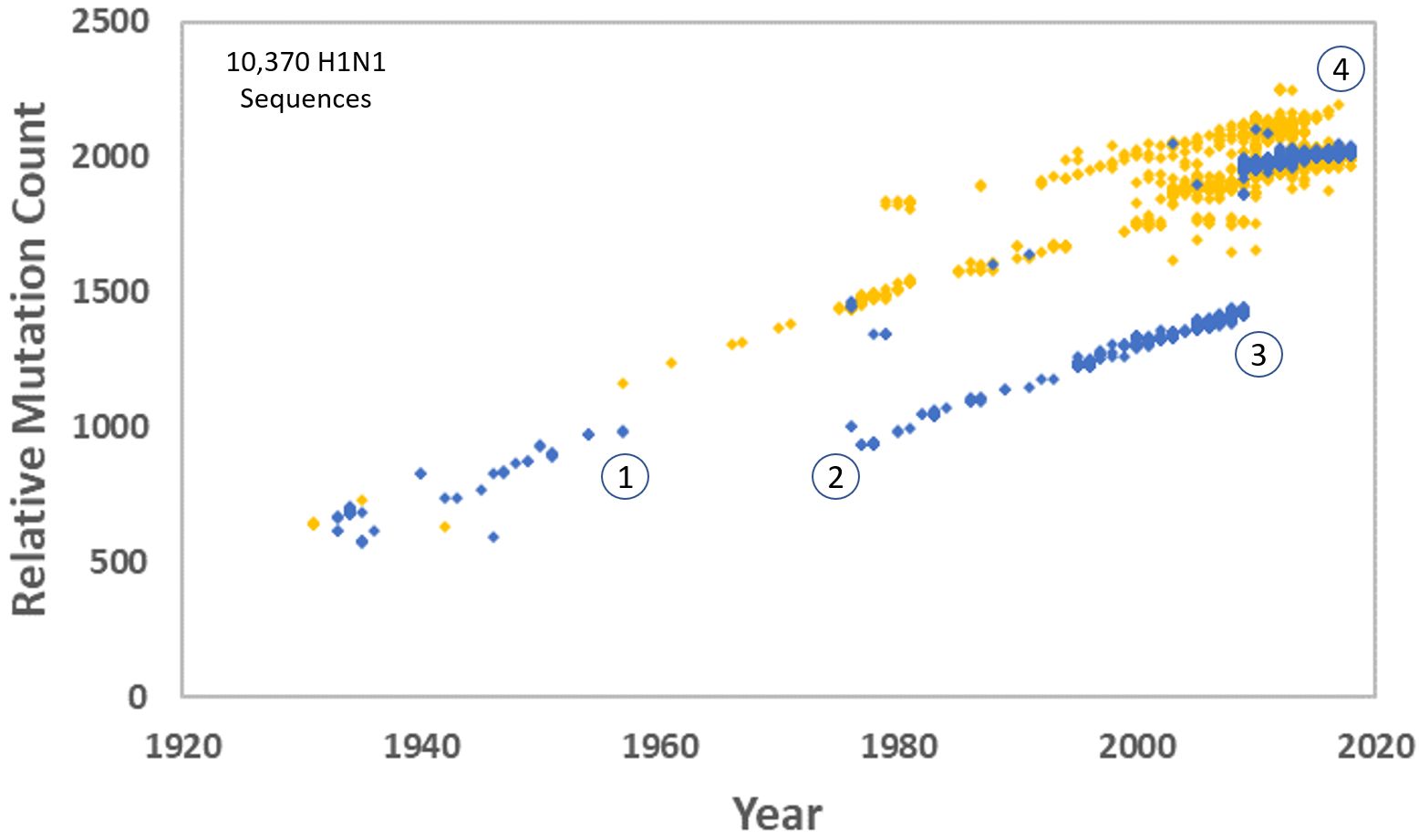
Every available H1N1 virus that had infected both human and swine as of 18 October 2018 was retrieved from the Influenza Research Database.3 Some sequences had to be removed due to quality control issues. Specifically, sequences with many Ns (i.e. missing data) tended to also be more diverged. This indicated the existence of poor-quality data. I culled every viral genome with more than five Ns. Also, some sequences were incomplete or missing one or more genomic segments.
There was a group of six consecutively named sequences from patients in New Zealand who were co-infected with the swine and human versions in 2009. These came from a study that claimed to have discovered reassorted viruses (genetically recombined swine and human H1N1) in several patients, even though other studies of a similar nature had failed to detect any reassortment.4 They also claimed the reassorted viruses did not spread to any other person. No other such ‘hybrid’ version can be seen in the massive database of viral genomes, and they left no viral descendants, so they were removed from the analysis.
The final dataset included 10,372 complete genomes that spanned 100 years of sampling.
I created an alignment for each of the eight genomic segments, trimming any sequence before the start codon or after the final stop codon and manually adjusting for indels. These were concatenated and entered into a single FASTA file, one line for each viral genome, totalling 13,133 nucleotides each. Distance was calculated by tabulating the number of differences compared to the 1918 strain (figure 1). The A/Brevig Mission/1/1918 strain is used as a reference for segments 1–3 and 5–8. The A/South Carolina/1/18 strain was used as a reference for segment 4 (see reference 1 for details). Occasional gaps and a few places where the sequence included an ambiguous letter call (e.g. an ‘S’ in a sequence indicated the letter was either a G or a C) were not counted as differences. There were no ambiguous calls in the reference sequence. Due to heavy sampling during specific years and in specific localities, there were many identical and near-identical sequences among the data. A pairwise comparison of all sequences was performed. If any pair differed by 10 or fewer nucleotides, the second member (alphabetically by strain name) of each pair was removed. This created a subset of 6,360 sequences with a minimum distance of 11 mutations. A neighbour-joining phylogenetic tree was created from a FASTA file of this subset in MEGA (version 7),5 then rooted to the 1918 strain (figure 2).

Results
As before, a clear break between the ‘human’ and ‘swine’ versions of the H1N1 virus can be seen (figure 1). This was caused by the accidental re-release of the virus in 1976 after being frozen in a sample dating back to approximately 1952.6 One can see occasional ‘swine’ H1N1 infections in humans over the years, and a huge burst after the 2009–2010 swine flu pandemic. In fact, from figure 1 it appears that all modern H1N1 infections in humans trace back to the swine version. It also suggests that the human version went extinct.
This is confirmed in figure 2, where we see that every H1N1 reported in humans since early 2009 traces back to the swine version. Indeed, the human version is clearly extinct after nearly a decade of non-reporting.
Discussion
Genetic entropy is supported by strong theoretical work, powerful numerical simulations, and real-world examples. The H1N1 virus is not immune to the effects of genetic entropy and we see major strains either weakening or disappearing entirely over time. This comes after decades of natural selection failing to remove thousands of incremental errors in the genome.1,2 Also, new viral lines have appeared suddenly, sometimes with significantly more mutations than comparable contemporaneous strains. These can be seen in the data presented here. This might be an example of the ‘mutator strain’ hypothesis of Carter, Lee, and Sanford.7 Or perhaps sudden mutation surges are part of the long-term history of the H1N1 virus, but this would make it even more prone to genetic entropy.
What is in store for the H1N1 virus? Over time, it should continue to accumulate mutations faster than selection can remove them. It should continue to become more and more attenuated and, barring an infusion of fresh genetic elements from a wild strain currently circulating in aquatic waterfowl, it should eventually meet its demise. Only the future can tell.
References and notes
- Carter, R.W. and Sanford J.C., A new look at an old virus: patterns of mutation accumulation in the human H1N1 influenza virus since 1918, Theor. Biol. Med. Model 9:42, 2012; tbiomed.com/content/9/1/42. Return to text.
- Carter, R.W., More evidence for the reality of genetic entropy, J. Creation 28(1):16–17, 2014. Return to text.
- Influenza Research Database, fludb.org. Return to text.
- Sonnberg, S., et al., Pandemic seasonal H1N1 reassortants recovered from patient material display a phenotype similar to that of the seasonal parent, J. Virology 90(17):7647–7656, 2016. Return to text.
- Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S., MEGA6: Molecular evolutionary genetics analysis version 6.0, Molecular Biology and Evolution 30:2725–2729, 2013. Return to text.
- Nakajima, K., Desselberger, U., and Palese, P., Recent human influenza A (H1N1) viruses are closely related genetically to strains isolated in 1950, Nature 274:334–339, 1978. See also reference 1. Return to text.
- Carter, R.W., Lee, S., and Sanford, J.C., Overview of the independent histories of the human Y-chromosome and the human mitochondrial chromosome; in: Whitmore, J.H. (Ed.), Proceedings of the Eighth International Conference on Creationism, Creation Science Fellowship, Pittsburgh, PA, pp. 200–216, 2018. Return to text.




Readers’ comments
Comments are automatically closed 14 days after publication.