

Journal of Creation 25(1):77–85, April 2011
Browse our latest digital issue Subscribe
The proportion of polypeptide chains which generate native folds—Part 1: analysis of reduced codon set experiments
Creationist scientists and Intelligent Design proponents have drawn attention to the sparseness of native-like folded proteins among random polypeptide sequences. Contrary to this opinion, it was alleged that protein folds are very common among random amino acid chains. We found this to be a surprising opinion, and this statement and the references cited prompted this six-part series. We review the best-known lines of evidence used to claim that random polypeptides often lead to native-like folds. We conclude that although a good estimate of the proportion of true native-like folds remains unknown, it is astronomically small. In Part 1 we address protein-folding experiments that are based on proteins constructed of very few but specified types of amino acids.
Many scientific reasons have been identified for doubting life could have arisen by natural processes.1–4 Examples include: only one enantiomer of amino acids must be used in most proteins; the difficulty of forming long protein chains in water; the statistical improbability of creating large RNA and DNA chains; the presence of the genetic code; and the need for many protein sequences which fold into a single stable conformation.
All these, and other, hurdles would need to be met with no intelligent guidance. If one prerequisite seems insurmountable, then the naturalist paradigm is implausible. In this six-part series we will evaluate one claim: that many random polypeptide sequences fold into a single stable conformation. The basis for these claims will be examined in depth in order to introduce some objectivity into the discussion. Of course, merely folding reliability is a necessary but not sufficient condition for important chemical processes. A particular fold only offers a stable scaffold upon which a useful geometric and electronic environment may or may not be available.
All known life-forms require a large number of different kinds of folded proteins together at the same time and location. If very few random sequences fold reliably, then obtaining such an ensemble naturally is very unlikely. This would indicate that abiogenesis beginning with proteins in the absence of a genetic code would have no scientific support.
Biological proteins are usually classified as fibrous, membrane and globular.5 Fibrous proteins are produced in the construction of hair, nails, tendons and ligaments. They are genetically expressed by mostly higher life-forms for structure but not for the fundamental biochemical processes necessary for life.
Membrane proteins probably make up the majority of all proteins found in the cell.6 They regulate, among many other functions, signal transduction, transport across the membrane and secretion. The structure of membrane proteins, however, is completely different when embedded in a membrane as to when in aqueous solution. This makes it very difficult to study and characterize membrane proteins in their relevant state. An authority pointed out recently that “Many proteins are retained within cell membranes and we know virtually nothing about the structures of these proteins and only slightly more about their functional roles.”7
Globular proteins have been the best studied, being easier to isolate in vitro, separated from other cellular bio-chemical interactions. Thousands of globular proteins are critically important, and are used as enzymes and for other purposes8. In fact, “Within all cells every reaction is regulated by the activity of enzymes.”9 To function at all, and reliably, globular proteins must fold into precise three dimensional structures. A polypeptide which produces a single, lowest energy folded structure will not thereby automatically provide any biological value, but such a scaffold is one prerequisite for useful function.
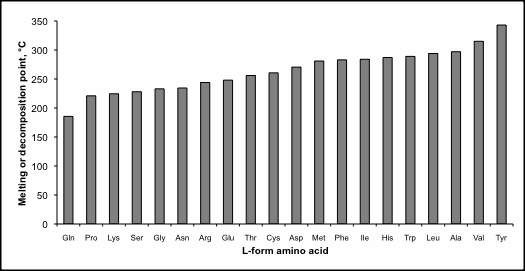
Table 1. Melting or decomposition (‘d’) temperature for biological l-amino acids.
| l-Amino acid | °C | |
|---|---|---|
| Ala | 297 | d |
| Arg | 244 | d |
| Asn | 234–235 | |
| Asp | 270–271 | |
| Cys | 260–261 | |
| Gln | 185–186 | d |
| Glu | 247–249 | |
| Gly | 233 | d |
| His | 287 | d |
| Ile | 284 | d |
| Leu | 293–295 | d |
| Lys | 224.5 | d |
| Met | 280–282 | d |
| Phe | 283 | d |
| Pro | 220–222 | d |
| Ser | 228 | d |
| Thr | 255–257 | d |
| Trp | 289 | d |
| Tyr | 342–344 | d |
| Val | 315 |
Amino acid polymerization is strongly disfavoured in water,10 rendering unreasonable the notion that life originated naturally in an oceanic “hot dilute soup”11 or warm pond12. Furthermore, the extreme high melting point of biological l-amino acids13 (see table 1 and figure 1) makes it essentially impossible to form linear chains under dry conditions. In fact, most natural amino acids decompose at high temperatures and don’t melt at all. Temperature in the 200–350°C range boils other organic substances and is inimical to life. In addition to a stable fold, globular proteins must remain soluble in water and not form tarry-like amorphous clumps.
In this series, we will be discussing the folding of globular proteins. Such folding must occur quickly and reliably into precise three-dimensional shapes to assure effective biochemical physiology. Therefore, if proteins arose naturalistically from random polypeptide sequences, the proportion of random chains which produce native-like folds is expected to be reasonably high. Contra this requirement, finding properly folded proteins among random sequences is rare.
Our evolution-touting counterparts are aware of the significance of this sparsity:
“This general argument has become of some importance as support for the view that proteins could not have arisen from natural pre-biotic chemical processes on earth and as support for creationism.”14
A key piece of evidence offered15 by Denton and co-authors for the view that properly folded proteins are common among random polypeptides was work carried out in Professor Sauer’s lab at MIT:
“In libraries of random amino acid sequences, alpha helical proteins displaying cooperative thermal denaturation of specific oligomeric states have been recovered at frequencies of 1%.”16
Sauer’s actual statement deviates in key details from the claim above:
“In fact, in libraries composed of random combinations of Leu, Gln, and Arg, proteins resistant to intracellular proteolysis were found at frequencies of about 1%. Purification and biochemical studies of several of these proteins revealed them to be α-helical, oligomeric, and to display reversible thermal denaturation. However, even the most native-like of these ‘random’ proteins differed from natural proteins in requiring some denaturant for solubility and in having extremely rapid rates of amide exchange.”17
Therefore the problem of random protein folding is not as simple as Denton et al. have suggested.
Testing a large number of random polypeptide sequences to determine how many produce native-like folds would be very difficult. One strategy is to build chains using only a few of the twenty natural amino acids, and to use smaller chains which are less likely to offer as many large random regions which interfere with folding. This reduces the variety of sequences which could be produced. Knowledge of what affects folding permits the research to be designed intelligently. For example, one of the strongest driving forces which cause folding is the ability to bury hydrophobic side chains inside the protein core, away from the aqueous environment.
A ‘binary pattern’ which uses a combination of hydrophobic (H) hydrophilic or polar (P) residues can facilitate the rough initial shaping of the protein. Analysis of existing α-helices in biological proteins shows a high frequency of residue patterns such as PHPPH and HPPHHH.18 For at least β-sheets located on protein surfaces (i.e. not buried into the protein core) a binary pattern such as HPHPH and PHPHP is often found.18 These ideas provide the insight to build polypeptides based on a reduced set of amino acids which satisfy the binary pattern as well as possible.
The experiment
The details of key experiments performed at MIT were published in 1994.19 What was done experimentally and what resulted?

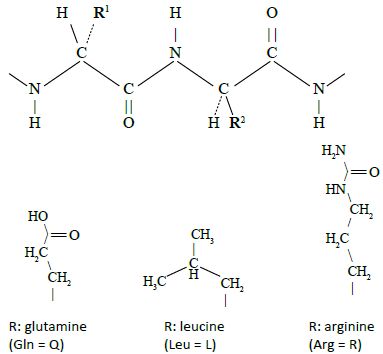
In Davidson and Sauers original work, a library of synthetic genes was prepared by linking together three or four oligonucleotide cassettes which consisted of 19 to 21 codons each. Each cassette was synthesized with a 10-bp (base-pair) self-annealing sequence at the 3’ end which allows synthesis of the second strand using special enzymes. This permits all the cassettes to be joined together as a single synthetic gene in a random manner giving rise to an exceptional repertoire of polypeptide sequences of around 80 residues composed predominantly of glutamine, leucine and arginine or ‘QLR’ proteins (figure 2).20 All the codons in the cassettes were based on the following formula: C (Cytosine) always in the first position; A (Adenine), T (Thymine) or G (Guanine) in the proportions 50%, 40% and 10%, respectively, in the second position; and A/G 50:50 in the third position. According to the rules of the genetic code, this will produce the amino acids glutamine (Q), leucine (L) and arginine (R) (figure 3). For experimental convenience, a tryptophan codon was added to permit fluorescent studies.
The joined cassettes were ligated to a backbone fragment which contained a promoter to ensure the gene would be expressed. At the carboxy-terminal tail the epitope tag DYKDDDDK was added; six codons for histidine to allow separation of the resulting protein by affinity purification; plasmid pBR322 origin of replication; an ampicillin-resistant gene and the lacIq gene. The backbone for this ensemble, the vector to be introduced into E. coli bacteria, was constructed from a plasmid (pDW239). The E. coli survivors from exposure to ampicillin permitted identification of those colonies which possessed the plasmid with the artificial gene, from which the expressed artificial proteins could be extracted.
Note that the authors do not claim in this paper19 that 1% of these artificial proteins fold properly: the quote above by Sauer alone was published two years later.17
Evaluation
We shall see that these experiments do not provide an estimate of the proportion of random sequences which would lead to a reliable native-type fold. What they do permit, however, is to prove that the proportion must be considerably lower than 1%! We shall now consider a series of corrective proportions, pn, that must now be applied to the 1% figure.
Correction p1: the proportion of the three QLR AAs (amino acids) is not random
Professor Sauer is an expert in protein chemistry and protein folding21 and knows which combination of residues, and in what proportion, would be best able to produce protein secondary structures. The propensity for a given amino acid residue to be found in α-helix22, β-strand23 or turn24 had been thoroughly documented25 long before Sauer and his co-workers performed this work. In 1996 he summarized26 the design elements appropriate to construct a small protein based on alpha helices (which knowledge he and his co-workers applied in the work described above19):
- “The use of residues with reasonable secondary structure propensities.” This is known from statistical studies using databases of secondary structures.
- “The choice of an appropriate binary pattern to ensure that polar residues face out to contact the solvent and non-polar residues face in to form a hydrophobic core.”
- “The use of turn and capping sequences to properly form the bend and terminate the helices.”
- “The use of hydrophobic side chains that allow complementary packing of the protein core.”
- “Perhaps the introduction of buried polar interactions.”

To optimize the chances of producing a protein with alpha helices, the particular codons described above in the cassettes were carefully designed to generate on average: 50% of the polar glutamine (Q); 40% of the hydrophobic leucine (L); and 10% of the charged residue arginine (R).20 The resulting polypeptides are referred to in the papers as QLR proteins. Notice how the carefully designed proteins generate an appropriate proportion between hydrophobic and hydrophilic residues; the use of 50% hydrophobic side chains neither too large nor too small; and a small amount of polar residues which could be buried into the protein core.
The sequences generated could incorporate Q, L or R randomly, affected only by the relative proportion of the codons used. This will allow many sequences to be generated which satisfy reasonably well binary patterning rules for helices. The experimental design also allows sequences to include the opposite, which is useful, since Marshall and Mayo note that,
“Most naturally occurring proteins contain some buried polar and exposed hydrophobic amino acid residues. In some cases, these residues are necessary for protein stability; for instance, many turns contain buried polar residues which form hydrogen bonds to main chain amides.”27
Of the library generated, three proteins were expressed sufficient for purification and studies.19 The proportion of Q/(Q+L) was found to vary between 0.456 and 0.534, as designed by the cassettes (Ibid.). The amount of amino acid ‘R’ varied from 7% to 13%. These ranges represent a small fraction of a percent of all sequences which could be generated using these three amino acids, and most of the excluded regions would not satisfy the intended ideal hydrophobic/hydrophilic proportions.
Therefore, the proportion of alpha helices and other features reported, which they believe have some semblance to proteins,19 (resistance to proteolysis and cooperative thermal denaturation) among all random QLR chains, can only be orders of magnitude smaller than the estimate reported. Incidentally, resistance to proteolysis need not imply native-like folded tertiary structure,28,29 neither must cooperative thermal denaturation.30
Correction p2: the AAs chosen easily form alpha helices
Davidson and Sauer admit that “The alpha-helical structure of the QLR proteins is not unexpected, given the high helical propensities of glutamine, leucine, and arginine.”31 The three proteins the authors isolated revealed extremely high fractional helicity values: 32%, 60% and 70%, which is not representative of random polypeptides. Hence, the extrapolation to random sequences using all natural amino acids will require a considerable correction factor.
Correction p3: the proteins were not soluble
A high concentration of chaotropic agents32 was needed to force the proteins which were isolated to remain in solution in water. Now, to be biologically useful, a protein should not form insoluble clumps as evidenced in this experiment.
We are not arguing that solubility is absolutely necessary to define true folding. However, together with other characteristics mentioned below, what was reported does not resemble native-like folds, and implies that these proteins are, to a high degree, amorphous conglomerates. Of all QLR proteins which could be formed, only the subset which displays the kinds of solubility expected for real, native-like folds (and none were found!) should have been factored into the calculations. Sticky hydrophobic patches must be avoided and p3 must have a value less than one.
Correction p4: the proteins were much too rigid
The authors point out that “None of the proteins showed significant loss of alpha-helical content up to 90°C, the highest temperature tested.”28 This is significant, as the authors point out: “We know of no natural proteins that retain their secondary structure in the presence of 6.0 M Gdn-HCl at 90°C.”33 Proteins require enough flexibility to interact with other partners to fulfil various functions. Structures which could be considered ‘evolutionary dead ends’ should not be counted as sequences with native-like properties. The significance of this observation will be revisited below. Once again, the value of p4 must be considerably less than 1.
Correction p5: the proteins lack conformational specificity
Native folds possess discrete conformation.
“In order to exhibit conformational specificity, a protein must satisfy three criteria. First, the protein must fold to a unique tertiary structure rather than exhibiting the conformational heterogeneity that is characteristic of molten globule and gemisch states. The protein must possess the desired oligomerization state … Finally the designed variants must assume the target fold rather than assuming an alternate fold.”34
NMR spectra can be very helpful in determining conformational states.35 But this was not performed in these experiments.
However, gel filtration experiments using the three isolated QLR proteins revealed that they exist as multimers. In other words, the QLR polymers generated adhere together. Filtration of artificial protein QLR-1 did not produce a homogeneous entity, and probably forms two or more oligomeric species; QLR-2 was probably a trimer and QLR3 a tetradecamer.36
The observations identified under p3–p5 suggest that the proteins are much too hydrophobic to produce native-like folds. Abnormally long alpha helices are formed, which become so stable, due to intra-molecular and intermolecular hydrophobic interactions, that as soon as one conformation has been reached, the protein is committed and cannot unfold and search for another slightly lower energy state. In other words, a large number of similar clumps of protein can form. Once the hydrophobic portions of long patches of alpha coils between two QLR members interact, somewhere along their chains they will remain strongly bonded and insoluble. This will prevent them from dissociating and attempt to realign in other, potentially more stable arrangements. The energy necessary to attain the dissociation transition state would be too high.
As a rule, real proteins must not be able to fold and lock into a multitude of similar conformations. Most of the wrong conformations would not interact correctly with the necessary chemical partners and could cause much damage by adhering where they should not. There are a few exceptions where natural proteins have been carefully designed to be able to equilibrate into more than one discrete and well-defined fold for special reasons, for example to serve as switches. In these cases it must be easy to alternate between intended conformations.
Since the necessary ‘N-cap’ and ‘C-cap’ sequences to clearly define where an α-helix begins and terminates are missing, a wide variety of alternative QLR protein conformations could form, contra what defines native-folded states. Blundell and Zhu37, and Richardson and Richardson38 have shown that special sequences are required to produce N-caps and C-caps.
Marshall and Mayo also point out that “sequences that are overly hydrophobic are prone to aggregation and are predicted to have a smaller energy gap between a target structure and alternate state.”39 Furthermore, “aggregates may arise from partially folded states rather than the native state.”40 One purpose of turns is to ensure that the relevant portions of the protein are held in the correct positions with respect to each other. Since no sequences which could be classified as turns were found, then a corrective factor much smaller than one is needed.
Correction p6: the data is based on only small proteins
The experiments were designed to produce QLR proteins about 60 or 81 residues long. We saw that these led to high α-helix content and amorphous structures which are much too stable to generate discrete folds. But the problems for these small domains, such as excessive stability, will be considerably worse for much larger and typical-sized domains41 of about 150 residues (and many folds exist which have more than 500 residues).42 For example, suppose the alpha coil were to become 25% larger for these polypeptides. The strength of the intermolecular interactions between multiple members would grow exponentially, and so would the possible locations along the helices where they could interact, leading to ever more, and stronger, ‘clumped’ variants.
There are far more QLR varieties which are 150 residues long than for the short 70 residue chains: 3150 / 370 = 1038, and in this vastly greater set the proportion able to fold properly must be considerably smaller than whatever the correct value for the smaller QLR proteins.
Correction p7: the data is based on only single-domain proteins
Most proteins have more than one domain, unlike those reported in the study.19 Each domain must fold properly and in the presence of the others.
Different domains interact with their intended biochemical partners, which permits the simplest life-forms to execute the necessary biochemical process with only a few hundred kinds of proteins.43 No life-forms are known which use only proteins of size 300 AA or less, and if tiny proteins of 60–81 residues are supposed to be the starting point for an evolutionary processes, then far more of them would be needed than using complex multi-functional versions.
Correction p8: the QLR proteins only generated alpha helices
Estimates vary for how many protein folds exist or could exist, but a value between one thousand and ten thousand is reasonable. Folds are classified in the CATH44 and SCOP45 databases, and FSSP/DALI46 classifications are also used to analyze possible protein structures. Folds are defined by alpha helices and beta sheets plus their geometric relationships together.
None of the folds which require beta sheets, nor mixtures of α + β, were generated in the experiments reported. The proportion of random polypeptides which possess different classes of secondary structure (α coils, β sheets and turns) needs to be estimated. The proportion leading to only α coils cannot be simply used as representative for the variety of folds found in nature.
Correction p9: the proteins are not forming native-type folds
A surprising observation is the inconsistent and misleading use of the words ‘protein fold’. Based on several facts, it is apparent that the CATH classification of ‘fold’ is meant when quoting the supposed 1% proportion of folded to non-folded random polypeptides. In CATH, this term has a precise meaning and is based on well-defined properties of biological proteins. For example, the figure shown in a recent paper15 by Dr Denton refers to “structural classes of protein folds” and references a book published by Orengo et al. who created and now maintains the CATH system. Furthermore, the same figure that was shown15 in that paper is used routinely in CATH literature.47
Denton’s paper quotes Sauer’s work, which we are evaluating here, referring to these proteins as being ‘folded’. But no CATH fold was obtained from the QLR proteins and Sauer and his co-authors never claimed so. If millions or billions of times more QLR sequences were to be generated then a true CATH-like fold might be found. Clearly corrective factor p9 is much smaller than one.
Discussion
Here, we have carefully analyzed whether protein folds are very common among random amino acid chains as it is claimed in the evolutionary literature. What we have found is quite revealing. Using CATH topology, the standard for protein folds, none of the randomly formed QLR proteins qualify as protein folds. The work reported by Sauer’s group was also very clear about this.18 They use the word ‘folding’ in a vague, non-technical way. The QLR proteins have one or more huge alpha helices, as deliberately designed, which then ‘clump’ together in an amorphous manner. A sheet of paper can also be crumbled in many ball-like amorphous ways with no resemblance to a protein. Native-like globular proteins fold into a precise discrete topology relying on secondary structures and other precise chemical interactions. The researchers themselves did not claim that the QLR polypeptides do this,18 nor was any CATH fold claimed for any of the polypeptides generated.
Although the above nine corrective terms are not independent, all of them undoubtedly have values far lower than one. Therefore, to extrapolate from Sauer’s QLR experiments to real folds based on the twenty natural amino acids of an average chain length, we would need to make some corrections:
Proportion of proteins able to fold p = 0.01 × p1 × p2 × p3 × p4 × p5 × p6 × p7 × p8 × p9.
Although we still cannot make a very good estimate for the proportion able to fold properly, the value of 1% is too high by many orders of magnitude. Considering the estimated pi values throughout the text, this proportion is expected to be very small.
The author contacted48 Dr Sauer hoping to discuss the above corrective factors and included the following comments to him to illustrate how 1% could not possibly be representative of typical domains nor proteins.
“Unfortunately, I cannot estimate very reasonably the proportion of random sequences based on the twenty optically pure amino acids, but lower limits can be placed. For example, all combinations of binary patterning49 for helices and coils would be covered by a pattern such as: (p/n)AA(p/n)AA …
“where p=polar; n=non-polar; A=Anything. This implies for a 150 residue domain that only every third position would pose a constraint. We’ll pretend any of nine out of twenty residues could be used as p or n. Once a polar or non-polar position is established, it is obvious from chemical considerations that every combination of AA in the next two positions, as I’ve permitted, would not really be possible (we would permit two and even three prolines next to each other; up to three huge tryptophans, one being part of the secondary structure, etc.), so our estimate is clearly far too generous. Even for turns, the pattern (p/n)AA is generous.
“Therefore, a lower limit everyone can agree to would be no larger than (9/20)51 = 10–18, which is not compatible with the 1% estimate.”
Note that these generous assumptions would imply a proportion of (9/20)100 = 10–35 for an assumed average size 300 AA protein.
This shows Denton’s 1% claim is an overestimation by mega-orders of magnitude. Sauer did not defend the 1% claim, but referred the author to unrelated work by Professor Szostak. This later work will be addressed in parts five and six of this series. Since Sauer’s pioneering experiments provide no guidance to quantitative native-like folding behaviour it is pointless to quote the 1% value, as done by Denton15, without clarifying its meaning. Watters and Baker also examined Sauer’s experiment and reached the same conclusion: “Few, if any, of the proteins in these screens were truly native-like, suggesting de novo formation of proteins may be very difficult.”50
Additional requirements to produce native protein folds

|

|
|
Figure 5. Examples of different folded topologies, displayed with RasTop 2.2. Left: CATH topology (fold) 3.40.5 (Ribosomal Protein L9; domain 1); Domain: 2hbaA00. Right: CATH topology (fold) 3.75.10 (L-arginine/glycine Amidinotransferase; Chain A) Domain: 1xknA00. | |
All known folds contain secondary structures: alpha helices and beta sheets, connected by chains called turns. The structure of the folded proteins can differ dramatically (see figure 5 for examples).51–53

|

|
|
Figure 6. Examples of two domains classified in the same topology, CATH fold 3.40.5. Displayed with RasTop 2.2. Left: Classification: 3.40.5.10; protein: Ribosomal Protein L9 domain 1; domain: 2hbaA00. Right: Classification: 3.40.5.20; protein: PriA/YqbF domain; domain: 2hjqA01. | |
The folds take only the backbone features into account, and on this basis commonalities can often be discerned for protein classified in the same fold (see figure 6).54,55 Although all proteins have been classified into only about a thousand unique folds in the CATH56 and SCOP57 databases, merely possessing helices or sheets is by no means sufficient to guarantee proper folding. Some additional relevant details includes:
- The non-α-helix22 and β-sheet23 portions of domains are involved in helping the folding process in bringing the
secondary structures together, which will then form into a suitable location. In some proteins the proportion of residues found in the turn can exceed 30% and are not random
structures58 Some features in protein turns have been identified and formally classified24:
α-turns59; β-turns60 (at least eight forms have
been identified)61; γ-turns62 (there are two
forms)63; π-turns64;
hairpins65; and ω-loops66.
The residues located in the turns bend the main-chain62 and affect the distance separating the secondary structures. Often specific motifs are found across members of some protein families, like the ‘tyrosine corner’ in Greek key beta-barrel proteins.62
The need for these special features to form the α-helix and β-sheet structures is one reason why so many amino acids are needed for biological proteins, and why the three used to construct the QLR proteins would not suffice. - To place α-helices at the correct location and to define their size, certain combinations of residues determine where they begin and end.67 For example, a four-residue ‘N-cap’ often terminates the end of helices and, in addition, often the next two residues which flank the box display typical hydrophobic interactions.
- Classification of folds is based for the most part on only the backbone topology, which is a minor portion of the protein mass. The side chains are
the key to providing biological function and also play important roles in determining whether a stable fold will be produced. The common use of the ‘cartoon’
representations of secondary structure leads to an over emphasis of the backbone. Figure 7 shows a typical ‘cartoon’-type display (left), in which the details of the
side chain are excluded, and the same structure with the side-chains included (right).
The side chains, it must be remembered, can interact whether attached to helices or sheet (see figure 7). And the details of the side chains are fundamental to understanding how a protein folds (and functions). These create micro-environments with precise spatial and electronic details. Therefore, structural and electronic requirements in the protein core place constraints as to which residues can be used at various residue locations. The side chains must ensure that in the core, cavities are avoided and strain due to interference between side chains must be minimized.
Only some combinations of amino acids are able to provide the environment necessary, through their side-chains, to produce stable conformations within a given fold. - Although proteins are generally dynamic, flexible structures, they must usually equilibrate most of the time near the native state. But this means that other conformations, which are energetically similar and easily attained, must often be prevented for a protein to be useful. Judicious choice of residues at specific positions can destabilize undesirable competitive conformations, and is one reason for intolerance to substitution by other residues with side chains which are quite different in size, shape, or charge characteristics.18

|

|
|
Figure 7. Ribosomal Protein L9 domain 1; domain: 2hbaA00, displayed with RasTop 2.2. Left: Usual ‘cartoon’ representation of secondary structures, with no side chains shown. Right: The same domain and orientation, including side chains. The backbone is shown as a solid chain. | |
Summary
The QLR proteins were expertly designed, knowing that otherwise it would be unlikely to find native-like folds among random sequences using 20 AAs. Although it is legitimate to first simplify a problem to gain insights, any quantitative conclusions made for the original problem cannot be based merely on the simplified work without reasonable extrapolations or experimental calibrations. We have not directly addressed all the papers dealing with QLR and other simplified proteins, but have shown that the claim that 1% of random proteins would produce native-like folds is unsupportable based on the QLR experiments.
References
- Truman, R. and Heisig, M., Protein families: chance or design, TJ (J. Creation) 15(3):115–127, 2001. Return to text.
- Behe, M.J., Dembski, W.A. and Meyer, S.C., Science and Evidence for Design in the Universe, Ignatius, San Francisco, CA, 2000. Return to text.
- Behe, M., Experimental support for regarding functional classes of proteins to be highly isolated from each other; in: Buell J. and Hearns, G. (Eds.), Darwinism: Science of Philosophy? Haughton Publishers, Dallas, TX, pp. 60–71, 1994. Return to text.
- Yockey, H.P., A prescription which predicts functionally equivalent residues at given sites in protein sequences, J. Theor. Biol. 67(3):337–343, 1977. Yockey, H.P., Information Theory and Molecular Biology, Cambridge University Press, 1992. Return to text.
- Whitford, D., Proteins: Structures and Functions, John Wiley & Sons, The Atrium, Southern Gate, Chichester, West Sussex, England, 2008. See Preface p. xiii. Return to text.
- Whitford, ref. 5, p. 85. Return to text.
- Whitford, ref. 5, p. 9. Return to text.
- Whitford, ref. 5, p. 189: “Enzymes catalyse metabolic reactions and in their absence reactions proceed at kinetically insignificant rates incompatible with living, dynamic, systems. The presence of enzymes results in reactions whose rates may be enhanced (catalysed) by factors of 1015 although enhancements in the range 103–109 are more typical. Enzymes participate in the catalysis of many cellular processes random from carbohydrate, amino acid and lipid synthesis, their breakdown or catabolic reactions, DNA repair and replication, transmission of stimuli through neurones, programmed cell death or apoptosis, the blood clotting cascade reactions, the degradation of proteins, and the export and import of proteins across membranes.” Return to text.
- Whitford, ref. 5, p. 189. Return to text.
- Sarfati, J.D., Origin of life: the polymerization problem, CEN Tech. J. (J. Creation) 12(3):281–284, 1998; creation.com/polymer. Return to text.
- en.wikipedia.org/wiki/Abiogenesis. Return to text.
- www.daviddarling.info/encyclopedia/D/DarwinC.html. Darwin speculated, in a letter to the botanist Joseph Hooker (1871), on the possibility of a chemical origin for life: “It is often said that all the conditions for the first production of a living organism are present, which could ever have been present. But if (and Oh! what a big if!) we could conceive in some warm little pond, with all sorts of ammonia and phosphoric salts, light, heat, electricity, etc., present, that a protein compound was chemically formed ready to undergo still more complex changes, at the present day such matter would be instantly devoured or absorbed, which would not have been the case before living creatures were formed.” Return to text.
- www.ajinomoto.co.jp/kfb/amino/e_aminoscience/ BC/b-4.html. Return to text.
- Lau, K. F. and Dill, K.A., Theory for protein mutability and biogenesis, Proc. Natl. Acad. Sci. USA 87:638–642, 1990; quote from p. 641. Return to text.
- Denton, M.J., Marshall, C.J. and Legge, M., The Protein Folds as Platonic Forms: New Support for the Pre-Darwinian Conception of Evolution by Natural Law, J. Theor. Biol. 219:325–342, 2002. Return to text.
- Denton, ref. 15, p. 336. Return to text.
- Sauer, R.T., Protein folding from a combinatorial perspective, Folding and Design 1(2):R27 (1996); p. R29. Return to text.
- Cordes et al., ref. 21, p. 5. Return to text.
- Davidson, A.R. and Sauer, R.T., Folded proteins occur frequently in libraries of random amino acid sequences, Proc. Natl Acd. Sci. USA 91:2146, 1994. Return to text.
- Davidson and Sauer, ref. 19, p. 2147. Return to text.
- Cordes, M.H.J., Davidson, A.L. and Sauer, R.T., Sequence space, folding and protein design, Current Opinion in Structural Biology 6:3–10, 1996. Return to text.
- en.wikipedia.org/wiki/Alpha_helix. Return to text.
- en.wikipedia.org/wiki/Beta_sheet. Return to text.
- en.wikipedia.org/wiki/Turn_(biochemistry). Return to text.
- Chou, P.Y. and Fasan, G.D., Ann. Rev. Biochem 47:251–276, 1978; Wilmot, C.M. and Thornton, J.M., J. Mol. Biol. 203:221–232, 1988. See also the table in Whitford, ref. 5 p. 182. Return to text.
- Cordes et al., ref. 21, p. 3. Return to text.
- Marshall, S.A. and Mayo, S.L., Achieving Stability and Conformational Specificity in Designed Proteins via Binary Patterning, J. Mol. Biol. 305:619–631, 2001; p. 621. Return to text.
- Tsuji, T., Kobayashi, K. and Yanagawa, H., Permutation of modules or secondary structure units creates proteins with basal enzymatic properties, FEBS Letters 453:145, 1999. Return to text.
- Matsuura, T., Ernst, A. and Plueckthun, A., Construction and characterization of secondary structure modules, Protein Sci. 11:2631, 2002. Return to text.
- Blanco, F.J., Angrand, I. and Serrano, L., Exploring the conformational properties of the sequence space between two proteins with different folds: an experimental study. J. Mol. Biol. 285:741, 1999. Return to text.
- Davidson and Sauer, ref. 19, p. 2148. Return to text.
- Chaotropic agents have the ability to destabilize hydrogen bonding and hydrophobic interactions. The authors used substances such as Gdn-HCl and urea. Return to text.
- Davidson and Sauer, ref. 19, p. 2150. Return to text.
- Marshall and Mayo, ref. 27, p. 623. Return to text.
- Marshall and Mayo, ref. 27, p. 624. “Well-folded proteins have relatively narrow linewidths in 1D 1H NMR spectra, while conformational heterogeneity and increased internal mobility at the millisecond to microsecond time scale, which characterize molten globule and aggregated states, result in broad and/or heterogeneous linewidths. Spectra of well-folded proteins are also characterized by pronounced chemical shift dispersion, which arises from the variety of unique magnetic environments that are present in a well-folded protein.” See their fig. 9 for examples. Return to text.
- Davidson, ref. 19, p. 2149. Return to text.
- Blumdell, T.L. and Zhu, Z.Y., The α-helix as seen from the protein tertiary structure: A 3-D structural classification, Biophys. Chem. 55:167, 1995. Return to text.
- Richardson, J.S. and Richardson, D.C., Amino acid preferences for specific locations at the end of α helices, Science 240:1648, 1988. Return to text.
- Marshall and Mayo, ref. 27, p. 626. Return to text.
- Marshall and Mayo, ref. 27, p. 627. Return to text.
- The average size of a globular domain, according to the CATH database, is 153 residues: Shen, M-y., Davis, F.P, and Sali, A., The optimal size of a globular protein domain: A simple sphere-packing model, Chemical Physics Letters 405:224–228, 2005. Return to text.
- Some domain are very large. From the CATH database, www.cathdb.info/index.html, status 12 May 2010: 743 domains in total with 500 or more AAs were reported, representing 18 unique domain folds, not counting presumed homologies. The largest reported domain is 1u6gC00 with 1146 AA. Some domain are very small. 27 domains total with 20 or less AA were reported, representing 7 unique domain folds, not counting presumed homologies. The smallest reported domain is 3cj8A01 with 13 AA. Return to text.
- Glass, J.I., Assad-Garcia, N., Alperovich, N., Yooseph, S., Lewis, M.R., Maruf, M., Hutchison III, C.A., Smith, H.O. and Venter, J.C., Essential genes of a minimal bacterium, PNAS 103(2):425–430, 2006. Return to text.
- www.cathdb.info/index.html. Return to text.
- scop.mrc-lmb.cam.ac.uk/scop/index.html. Return to text.
- ekhidna.biocenter.helsinki.fi/dali. Return to text.
- Orengo, C.A. Michie, AD, Jones, S. et al., CATH—a hierarchic classification of protein domain structures, Structure 5(8):1094, 1997; www.cell.com/structure/retrieve/pii/S0969212697002608. Return to text.
- Robert Sauer, e-mail message to author, 5 June 2010. Return to text.
- West, M.W and Hecht, M.H., Binary patterning of polar and nonpolar amino acids in the sequences and structures of native proteins, Protein Sci. 4:2032, 1995. Return to text.
- Watters, A. L, and Baer, S., Searching for folded proteins in vitro and in silico, Eur. J. Biochem. 271:1615–1622, 2004. Return to text.
- www.geneinfinity.org/rastop/. Return to text.
- www.cathdb.info/cathnode/3.40.5. Return to text.
- www.cathdb.info/cathnode/3.75.10. Return to text.
- www.cathdb.info/domain/2hbaA00. Return to text.
- www.cathdb.info/domain/2hjqA01. Return to text.
- www.cathdb.info/index.html. Return to text.
- scop.mrc-lmb.cam.ac.uk/scop/index.html. Return to text.
- Whitford, ref. 5, p. 47. Return to text.
- An α-turn is characterized by hydrogen bond(s) in which the donor and acceptor residues are separated by four residues. Wikipedia, ref. 24. Return to text.
- A β-turn (the most common form) is characterized by hydrogen bond(s) in which the donor and acceptor residues are separated by three residues. Wikipedia, ref. 24. Return to text.
- Depending mainly on whether a cis isomer of a peptide bond is involved and on the dihedral angles of the central two residues. Wikipedia, ref. 24. Return to text.
- A γ-turn is characterized by hydrogen bond(s) in which the donor and acceptor residues are separated by two residues. Wikipedia, ref. 24. Return to text.
- γ-turn has two forms a classical form with (φ, ψ) dihedral angles of roughly (75°,-65°) and an inverse form with dihedral angles (-75°, 65°). Wikipedia, ref. 24. Return to text.
- A π-turn is characterized by hydrogen bond(s) in which the donor and acceptor residues are separated by five residues. Wikipedia, ref. 24. Return to text.
- A hairpin is a special case of a turn, in which the direction of the protein backbone reverses and the flanking secondary structure elements interact. Wikipedia, ref. 24. Return to text.
- An ω-loop is a catch-all term for a longer loop with no internal hydrogen bonding. Wikipedia, ref. 24. Return to text.
- Cordes et al., ref. 21, p. 4. Return to text.
Readers’ comments
Comments are automatically closed 14 days after publication.