

Journal of Creation 27(3):84–92, December 2013
Browse our latest digital issue Subscribe
Incomplete lineage sorting and other ‘rogue’ data fell the tree of life
The ‘tree of life’ (TOL) popularized by Darwin and used as the inferred pattern of life’s history is the centrepiece of evolutionary biology. The molecular genetics revolution has presented many contradictions for the TOL and the modern Darwinian synthesis. Incomplete lineage sorting (ILS) is a discordant and pervasive outcome produced when constructing phylogenetic trees using homologous biological sequence data across all types of life studied. The ILS paradigm is characterized by segments of DNA that produce phylogenetic trees with different topologies compared to hypothetical inferred evolutionary trees. While ILS within closely related taxonomic groups can largely be explained by horizontal genetic variation and limitations on accurately sampling large populations, ILS across clearly different and unrelated kinds of organisms represents a mosaic of DNA sequence patterns that cannot be explained by common ancestry. Other ‘rogue’ genetic data that defy the TOL are microRNA genes and taxonomically restricted genes. MicroRNAs produce completely different trees compared to other gene sequences and appear unexpectedly in taxa. Taxonomically restricted genes also appear abruptly without evolutionary precursors, lack homology to other genes, and uniquely define taxon. Genetics research consistently reveals patterns of DNA mosaics that defy evolution and vindicate biblical creation ‘after their kinds’.

The dominant metaphor of evolutionary biology is the overall concept of a branching tree described by Darwin in 1859, in his book titled On the Origin of Species by Means of Natural Selection, or The Preservation of Favored Races in the Struggle for Life.1 Using a single illustration of a tree diagram with branching patterns and calculations, Darwin illustrated the gradualistic divergence of species over time (figure 1). However, from the time of Darwin to the early molecular protein work of Zuckerkandl and Pauling in the 1960s, these trees were largely based on closely-related species and groups of organisms.2 For an example of a phylogenetic tree, see figure 2.
According to evolutionary theorists, the simple assumption of phylogenetics and the development of evolutionary trees from biological sequence implies that “as the time increases since two sequences diverged from their last common ancestor, so does the number of differences between them, tree estimation seems to be a relatively simple exercise: count the number of differences between sequences and group those that are most similar”.3 Nevertheless, evolutionary biologists also recognize that “The simplicity of such an algorithm underestimates the complexity of the phylogenetic-inference problem”.3 In fact, the main problem with phylogenetic inference is that of discordant data. This rogue data provides no support for gradualistic Darwinian assumptions and the inferred common ancestry across the spectrum of life. In the case of phylogenetics, where certain homologous sequences across taxa exist and make possible the use of comparative techniques, the discordant data is typically referred to as incomplete lineage sorting or ILS. For a simple graphical example of ILS as displayed in phylogenetic trees, see figure 2.

Prior to the recent advent in DNA sequencing, a 1965 report by Throckmorton using morphological characters in the genus Drosophila (fruit fly) described how similarity in individual phenotypic traits did not consistently predict assumed evolutionary relationships when evaluated independently.4 Later, in 1978, Farris made one of the first attempts at using one of the early tools of molecular genetics (chromosome inversion data) to infer evolutionary phylogenies and ran into the same enigmatic issue of ILS.5 It should be noted, however, that Drosophila is an animal with large populations and short generation times. In interrelated and interfertile populations that may be largely separate, chromosome inversions that are tolerated will not completely inhibit gene flow.6 Thus, ILS among closely related taxa can largely be explained as a common feature of horizontal genetic variation within kinds as recently demonstrated among a tribe of cichlid fishes.7 The presence of ILS among closely related taxa is also affected by the fact that accurately sampling and characterizing large populations, such as cichlid fish in multiple lakes and rivers, can be very difficult.8

While the findings and reports of ILS within single taxa are noteworthy, they do not provide an adequate evolutionary explanation for many recent studies in which ILS is observed across completely unrelated kinds of organisms that are obviously not interfertile and have no flow of genetic information between them. Evolutionists like to extrapolate the observed variation within kinds and associated polymorphisms (sequence variations) inherent to horizontal genetic variability as an explanation for ILS among non-interfertile taxa. In fact, recent phylogenetic reports in the literature describing ILS will often cite these early papers based on variation within an interfertile group as an explanation for the discontinuity they are observing on a grand scale in the tree of life. However, these so-called ‘ancient ancestral polymorphisms’ must involve the transfer of ILS-related fragments across skipped taxa in a lineage—a feat which is impossible for non-interfertile taxa (see figure 3 for an illustration).
As molecular phylogenetics became more advanced and distinctly different kinds of organisms across the spectrum of life were being compared, ILS was becoming a major issue for evolutionists. In a 1979 study by molecular phylogenetics pioneer Felsenstein, it was acknowledged that the evolutionary enigma of ILS was not only going to be a common caveat in phylogenetics research, but would also be especially problematic among studies where multiple types of traditional and new molecular technologies were used in combination.9 As stated in a 2006 phylogenetics review by Maddison and Knowles: “It is now well known that incomplete lineage sorting can cause serious difficulties for phylogenetic inference”.10 McCormack et al. reiterated this sentiment in 2009 stating that “gene trees can disagree in topology with the species tree that contains them. Incomplete lineage sorting is one of the most common reasons in nature for this discord”, and “Discord among gene trees poses a serious challenge for phylogenetic estimation”.11
As predicted by Felsenstein, the problem of discordant evolutionary trees did not disappear with the advance of molecular technologies that allowed for increased amounts of sequence determination for proteins, DNA, and RNA. In fact, the problems for phylogenetics got worse and continue to persist and plague evolutionary presuppositions. In a recent large-scale phylogenomic study across 36 mammalian genomes by Boussau et al., the authors note, “In the case of the mammalian phylogeny, the role of ILS seems particularly problematic”.12 In addition, the large increase in genomic data has not helped, but actually clouded the tree of life as noted in a recent paper by Degnan and Rosenberg, who stated: “Recent advances in genealogical modelling suggest that resolving close species relationships is not quite as simple as applying more data to the problem”.13
Evolutionists attempt to explain the presence of ILS across major taxon by appealing to the shuffling of genetic variation that occurs within taxa or what creationists would define as created kinds. This appeal to micro-evolution or mechanisms associated with horizontal variation as an explanation for ILS is a common theme referred to as ‘ancestral polymorphisms’. The idea is that the DNA segments that confer ILS in completely unrelated taxa are said to be the result of ancestral variation that occurred prior to the divergence of major lineages. However, as one recent study among placental mammals showed using just shared homologous DNA regions, numerous interbreedings between unrelated and non-interfertile species would have had to take place for this to occur.14 Furthermore, this idea contributes nothing to the evolutionary resolution of the numerous genomic regions across the diverse spectrum of life that show complete mosaics of DNA patterns.
A good example of how ILS plagues the evolutionary paradigm can be found in the genetic studies of human origins. Evolutionists have long maintained that modern primate species (including, in their view, humans) are branches on an evolutionary tree that lead back to a common ancestor. Of course, ILS and the problems it presents for primate evolutionists in regards to mosaics of morphological traits existed before the days of DNA sequencing and continues to cause much controversy.15
One of the first papers to expose the problem of DNA sequence ILS in the area of primate evolution was published in 2007 by Ebersberger et al.16 In this study, researchers used selected homologous sequences present in humans, chimpanzees, and gorillas in which they trimmed out the gaps, insertions, and deletions. Despite this optimization of the data, a majority of the trees showed high levels of incongruence compared to the inferred model of human evolution. The researchers stated:
“Thus, in two-thirds of the cases [trees], a genealogy results in which humans and chimpanzees are not each other’s closest genetic relatives. The corresponding genealogies are incongruent with the species tree. In concordance with the experimental evidences, this implies that there is no such thing as a unique evolutionary history of the human genome. Rather, it resembles a patchwork of individual regions following their own genealogy.”
The 2012 published report of the gorilla genome sequence added even more ILS data to the growing problem facing the primate evolution model.17 In a high proportion of cases, depending on which DNA fragment is used for the analysis, evolutionary trees based on DNA sequences showed that humans are more closely related to gorillas than chimpanzees. The overall outcome, as with previous studies, was that no consensus path of common ancestry between humans and various apes exists, and no coherent model of primate evolution could be achieved. The authors reported that “in 30% of the genome, gorilla is closer to human or chimpanzee than the latter are to each other”.
Masking the problem by pruning rogue data
If a naturalistic dogma of the origin of biological life was going to be successfully sold to the academic community in the new age of biotechnology, a solution to cloak and obfuscate the evolution-negating inconvenient data needed to be achieved. The response to this dilemma was undertaken, not by biologists, but by mathematicians such as Felsenstein and Kingman who essentially pushed the biological reality and inconvenience of ILS off to the side and used statistical models that manipulated the data by a combination of boot-strapping, subjective data selection, and injecting biased evolutionary presuppositions into the overall analysis methodology.9,18,19 The result was that the real meaning of so-called discordant or discontinuous data was mathematically smoothed over and manipulated to fit a preconceived Darwinian outcome.
Nevertheless, the ongoing inability of mathematicians to satisfy the nature of real world biology continues to grow as an increasingly glaring evolutionary problem and is exacerbated by the genomics revolution and the escalating amounts of discordant data. A 2006 article by Maddison and Knowles, aptly titled ‘Inferring phylogeny despite incomplete lineage sorting’ made the following statement in regard to the inability of prevalent mathematical models to deal with the exponential increase in so-called phylogenic anomalies: “Although phylogenetic patterns generated by incomplete lineage sorting have been discussed for many years, considerable work remains to develop and assess methods that consider these issues during phylogenetic reconstruction.”10
Despite the widespread use of various types of data-smoothing techniques, the mosaic pattern of life presents many difficulties for evolutionary scientists to overcome. In 1996, Wilkinson introduced the terms ‘rogue data’ and ‘rogue taxa’ to describe discordant biological sequence that negated the development of inferred evolutionary trees.20 This rogue data is typically given other terms like ‘ambiguous’ or it is considered to provide ‘insufficient phylogenetic signal’. In fact, the concept of developing bioinformatics filters to remove unruly ‘rogue’ data that does not produce favorable evolutionary outcomes has been an important goal of bioinformatics software developers as the genomics revolution progressed. In 2002, a filtering algorithm was developed that would assess evolutionarily unfavorable DNA that the authors defined as ‘phylogenetically discordant sequence (PDS)’ and eliminate them if they fell outside a pre-set similarity threshold.21 It is now very common to use various types of software programs that ‘prune’ (eliminate) insertions, deletions, and other types of discordant data to produce more favorable multiple sequence alignments and then evolutionary trees.22,23 Recently, software was developed and made available via web server interface that removes unruly non-evolutionary genomic sequence from your data set to give you what is termed the ‘true tree’, otherwise known as the inferred hypothetical phylogeny.24 The authors of this report state:
“When rogue taxa are identified based on support values that are drawn onto a best-known tree, we observe that pruning these rogues yields trees that are topologically closer to the true tree”.
Coalescence theory
Coalescence theory is a theoretical concept wherein the variants of a particular gene or DNA segment are hypothetically traced to a single ancestral gene copy in a so-called molecular genealogy.19 The theory postulates that the probability that two lineages coalesce in preceding generations in regard to DNA similarity is the probability that they share a common ancestor. It can also refer to a node in the evolutionary tree, known as the phylogeny. The coalescence in a macro-evolutionary context typically represents a hypothetical sequence or node placed in the tree.
One must also keep in mind that in this scenario, a single gene stands in proxy for each species or organism. The main problem with this idea is the fact that different genes and gene families can produce different evolutionary trees. Degnan and Salter noted this issue and stated the following in a 2005 report: “For the problem considered in this paper, only one gene is sampled per population, and intra-specific variation is not modeled”.25
Evolutionists explain these differences between genes by assuming varying rates of evolution in different parts of the genome and have noted how this can foul up any prediction of divergence, which is also the last hypothetical point of coalescence. Holder and Lewis state: “The rate of sequence evolution is not constant over time, so a simple measure of the genetic differences between sequences is not necessarily a reliable indication of when they diverged.”3
So how is some sort of collective whole genome coalescent achieved for multiple genes that individually, each produce their own different trees? This is a ubiquitous anti-evolutionary feature referred to by evolutionists as “massive amounts of incongruence in various data sets due to incomplete lineage sorting”?26
One method to resolve this thorny problem is the ‘concatenation approach’ where multiple sequences from each organism are concatenated to produce ‘supergene’ data and then analyzed with traditional approaches (maximum likelihood, maximum parsimony).27 Of course, this approach also involves the biased manual selection of only evolutionary favorable sequences (full of phylogenetic signals) and the trimming of gaps and flanking sequences that contain ‘rogue data’. A second method is to run multiple individual gene tree analyses, and derive the consensus sequence, sort of averaging things out into a single tree.28 A third approach has often been referred to as the ‘democratic vote’ method, which involves selecting the most commonly occurring gene tree out of many individual analyses.26
Finally, one of the most extensively investigated methodologies in recent years, and perhaps also the most theoretical and obfuscated, involves modelling the largely hypothetical coalescent process using a variety of statistical methodologies.10,29,30,31 It must be noted that these are ‘inference’ based methodologies and all the modern versions of this analysis essentially derive from an idea popularized by Maddison, in which he proposed minimizing deep coalescences (rogue anti-evolutionary nodes in the tree).32 In other words, hypothetical modelling and statistical optimization criterion are now the chief goals for inferring the politically correct evolutionary tree from ever-increasing sets of incongruent gene trees, thanks to the genomics revolution.
In what is considered a classic paper on phylogenetics and the pervasive issue of ILS at the dawn of the high-throughput genomics revolution in 1997, Maddison makes the following statement:32
“In considering these issues, one is provoked to reconsider precisely what is phylogeny. Perhaps it is misleading to view some gene trees as agreeing and other gene trees as disagreeing with the species tree; rather, all of the gene trees are part of the species tree, which can be visualized like a fuzzy statistical distribution, a cloud of gene histories. Alternatively, phylogeny might be (and has been) viewed not as a history of what happened, genetically, but as a history of what could have happened [emphasis added].”
This general idea promoted by Maddison and now widely pursued among most researchers now involves a combination of selecting only homologous regions between combined taxa, the extensive pruning of rogue data, and the testing and comparison of various hypothetical coalescent models based on the concept of ‘inference’—using the inferred hypothetical evolutionary tree as the gold standard. And as noted by a recent review paper, most recent efforts now largely ignore ILS as a chief cause of incongruence in their data analyses, although the researchers themselves still widely acknowledge that it pervasively exists.26
Ancestral sequence reconstruction
Using DNA and protein sequence data, hypothetical estimates of so-called ancestral gene sequences and population sizes can be derived. Despite the fact that these are purely hypothetical constructs and heavily based on evolutionary presuppositions, the conclusions are often touted as factual.
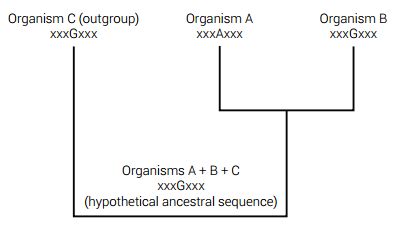
The construction of ancestral gene sequences has some validity within viral genetics related to epidemiology and also within closely related taxa.33 However, in such cases where an ancient hypothetical sequence is being ‘resurrected’ in a deep node in the phylogenetic grand tree of life, the issue is purely speculative no matter how much statistical theory is applied.34,35 For a visual depiction of ancestral sequence reconstruction, see figure 4.
At present, one of the more popular approaches is to take biological sequences from a multiple sequence alignment along with the resulting phylogenetic tree data as input to re-construct the hypothetical sequence in a process that can be divided into two parts. The first part reconstructs the individual characters, whether they are amino acids or nucleotides from known sequences. The second part is even more speculative and involves the reconstruction of postulated insertions and deletions. The results are said to provide the most ‘probable’ ancestral sequences in each node of the phylogeny—all based on the ‘probability’ that the evolutionary hypothesis (the gold standard) is true.33 This is a clear case of circular logic using evolution to prove evolution and manipulating the data accordingly.
While sequence substitutions are based on models of inferred evolution, determining insertions and deletions is even more problematic and speculative.36 Besides the highly speculative nature and naturalistic presuppositions, this whole scenario of ancestral sequence reconstruction is also clouded by the fact that different genes produce different phylogenies between and within taxa. How can any sort of ancestral sequence reconstruction be valid if the sequences themselves, when known, do not consistently perform as anticipated within a macro-evolutionary paradigm?
MicroRNA genes re-draw the tree of life
A coalition of palaeobiologists have been literally rewriting the tree of life in complete contradiction to the standard evolutionary paradigm using microRNA (miRNA) genes as opposed to selected sets of homologous protein coding genes shared among species.37,38,39 MicroRNAs are small regulatory RNA molecules (~22 bases) that bind to and regulate the transcripts of both protein coding genes and non-coding RNAs. Their activity has been implicated in virtually every biological process studied in plants and animals.40 In fact, scientists now believe that many eukaryotic RNA transcripts communicate through a new ‘language’ mediated by microRNA-binding sites called ‘microRNA response elements’ located in their three prime untranslated regions (3’ UTRs).41 These 3’ UTRs act like long regulatory tails on the ends of genes and can contain hundreds to thousands of built-in miRNA-controlled regulatory switches per gene RNA copy.42 Therefore, a completely different class of DNA sequences that are separate in their structure and function from conserved protein coding genes, but yet regulate them with a complex system of control, give completely different sets of phylogenetic trees.
The reason that evolutionists have proposed the use of miRNA sequence is because they note that miRNA genes “are continuously being added to animalian genomes, and, once integrated into a gene regulatory network, are strongly conserved in primary sequence and rarely secondarily lost, their evolutionary history can be accurately reconstructed”.43 Thus, constructing phylogenies with miRNAs is not only based on similarity of DNA sequence, but also on a presence or absence scenario. This whole paradigm in and of itself has caused concern among traditional evolutionists because these miRNAs appear suddenly in different kinds and groups of organisms without any evolutionary sequence precursor. This is in addition to the fact that they produce different evolutionary trees from the standard inferred ones.37
Plant and animal miRNAs exhibit significant differences in both their modes of biogenesis and their systems of regulation for their target mRNAs.44 MicroRNA gene sequences are also completely different between plants and animals and appear suddenly in each group—even showing no miRNA ancestry with the hypothesized crown ancestors of eukaryotes (protozoans).44 Even at the base of the metazoan (animal) tree, new groups of miRNAs appear suddenly without any trace of ancestry as in the case of a recent study which showed this trend in four different lineages of sponges—all contained completely novel classes of miRNAs.45 When their data was compared to other studies, the authors stated “we suggest that miRNAs evolved multiple times independently not only among eukaryotes, but even within animals.”
Among reptiles, miRNAs showed that turtles are more closely related to lizards than to birds or crocodiles which is the opposite of what evolutionists derived from datasets using other genomic sequences, including genes.46 In addition, the researchers stated that they found that turtles and lizards share four unique miRNA gene families that are not found in any other organisms’ genome. As with other animals, these novel miRNAs appeared suddenly with no trace of ancestral evolutionary beginnings.
In the case of mammals, phylogenies constructed with miRNAs for eight different taxa produced completely different trees than the standard inferred evolutionary trees. In fact, results from miRNA research are also completely contradicting data from a recent phylogenetic study published for mammals using 26 highly conserved gene fragments.47 Needless to say, the conflicting results are causing much controversy within the evolutionary community.
Gene landscape differences between taxa defy evolution
One of the key problems for performing phylogenetics on a genome scale is showing how entire networks of co-regulated genes somehow evolved together. One approach to studying this issue is the cross taxon analysis of co-regulated genes in gene neighbourhoods. Depending on the chromosome, a significant number of genes that operate within the same physiological network will also be located close together in the genome and have shared expression patterns. A recent study compared homologous gene neighbourhoods between a diverse group of animals (human, chimpanzee, mouse, rat, chicken, zebrafish, fruit fly).48 The results of the study showed how incongruence in phylogenies is in part, also based on the complete presence or absence of entire gene sequences across taxa for entire gene networks with similar functions. The authors of the study state:
“Surprisingly, the genes found in functional neighborhoods shared by different organisms are not necessarily orthologous. That is, when two species share functional neighborhoods, the genes forming these clusters may be different in each species. One might expect that if such functional neighbourhoods emerged in a particular period of the evolution and apparently were maintained since then (given that they are shared by all the descendant species), these clusters were essentially composed by ortholog genes. Nevertheless, this is not the case.”
Quite unexpectedly, these gene neighbourhoods with similar functions between taxa were populated by different genes. Instead of being conserved as predicted by evolution, these functional neighbourhoods contained a higher degree of synteny (gene order and/or absence) differences than other types of genes on average throughout the genome.48 In another study, researchers compared the genomic landscapes surrounding highly homologous genes between humans, chimpanzees and macaques. For 18% of the genes, it was found that large amounts of discontinuity in the DNA landscape existed, which they termed ‘altered gene neighbourhoods’.49 Despite the high levels of homology between the targeted genes among humans and apes, the genomic landscapes surrounding them was in many cases, taxon specific. Thus, merely looking at homologous genes is insufficient. The genomic context in which they are located and function is also important to consider, but often not supportive of macro-evolutionary models.
Taxonomically restricted genes
One sort of evolution-negating ‘rogue’ data of particular note is the ubiquitous occurrence of taxonomically restricted genes (TRGs; also called orphan genes) being discovered in the sequencing of all genomes. In a 2009 review on the subject, Khalturin et al. noted that, “Comparative genome analyses indicate that every taxonomic group so far studied contains 10–20% of genes that lack recognizable homologs in other species”.50 In another recent review Tautz and Domazet-Lošo state that “every evolutionary lineage harbours orphan genes that lack homologues in other lineages and whose evolutionary origin is only poorly understood”.51
TRGs were first discussed during the outcome of the yeast genome sequencing project which predicted that at least one third of the identified genes fell into this category, but is now believed to be about 11%.50,51 Comparative genomics has shown that TRGs are a universal feature of every animal genome.50,52 TRGs are thought to be particularly important for taxa-specific developmental adaptations and the interaction of the organism with the environment.51
Partitioning the genes discovered within a new genome into different taxonomically based categories is still being refined, although the category of TRGs is readily discerned by the fact that the genes are only found in that particular organism. In general, the demarcation of various levels of DNA sequence homology is based on threshold values of the alignment algorithm and can become quite involved, such as in the case of the extreme ecoresponsive genome of Daphnia pulex in which nine different categories were developed, one being TRGs.53 In a more simplified and general model, there appear to be three different classes of protein coding genes within eukaryotic organisms as illustrated by a recent study in zebrafish:
- Genes that are shared across broad groups of eukaryotes referred to as ‘evolutionarily conserved genes’.
- Genes that are only shared within a broad group of organisms (e.g. Teleost fishes).
- Genes that are specific to a certain interbreeding taxon or TRGs (e.g. zebrafish).52
TRGs appear suddenly in genomes with no evolutionary precursors and their presence has been a mystery explained only by imaginative scenarios with no hard proof of such a mechanism existing. The two main hypothetical propositions for TRGs is ‘de novo synthesis’ and ‘horizontal gene transfer’ (also referred to as lateral gene transfer).
The de novo synthesis scenario invokes the idea of genomic shuffling, which is central to the well-known evolutionary idea of ‘tinkering’. The evolutionary concept of tinkering is central to the paradigm of molecular evolutionists and was first coined and proposed in Francois Jacob’s famous treatise ‘Evolution and tinkering’.54 Because evolutionists attempt to invoke the duplication of genes as a method for the origination of most genes in the genome, TRGs don’t fit this model because they have no other sequences in the genome from which they could have been copied—they also by definition don’t have any paralogs (similar sequences in the same genome). Tautz and Domazet-Lošo attempt to invoke the idea of tinkering and even claim that six such genes found in various species prove it. However, this is merely a case of using evolution to prove evolution. The original sources describing these genes merely assumed (inferred) de novo synthesis simply because the sequences had no homologs. The concept of tinkering or random shuffling of DNA to produce a complex functional gene is purely a mythical process with no proof. In fact, it defies the very basic laws of probability and information recently covered in great detail by Meyer.55
Another idea that is invoked is horizontal gene transfer wherein the TRGs in one taxa originated by being transferred from another taxa through non-sexual means. Since by definition TRGs have no homologs in other taxa, the donor organism would be unknown. In bacteria, horizontal gene transfer does occur although its frequency has yet to be accurately determined through a variety of mechanisms unique to prokaryotes.56 While microbe-to-host transfer of genes has been documented in eukaryotes, the transfer of genes between metazoans lacks a viable proven mechanism.57 In the case of the recent determination of gene sequences in bdelloid rotifer (micro-invertebrate), researchers ascribed the presence of ~10% of the creature’s actively transcribed genes to horizontal gene transfer from eubacteria, fungi, protists, and algae without any evidence or documented mechanism for how they may have got there.58 The idea of horizontal gene transfer was simply invoked because the genes showed some level of similarity to genes in other taxa. In regards to rotifer TRGs, the researchers recorded over 61,000 gene sequences that were expressed from rotifers grown in stressed and non-stressed conditions and could only find sequence similarities with genes from other creatures for 28,922 sequences (less than half).
Summary
The use of molecular phylogenetics to reconstruct a genetic phylogeny from multiple alignments of DNA segments that are homologous, yet also diverse, is done with the goal of inferring macro-evolutionary history. In these types of studies, incongruities of the genetic comparisons are a very common problem. Major incongruence between gene trees is the main challenge faced by phylogeneticists in attempting to document macro-evolution. This occurs with similar genes between taxa. The type of gene or DNA sequence used also produces different results. So not only is there incongruence observed across taxa but also across gene types and/or DNA elements.
These disparities are referred to as incomplete lineage sorting (ILS), which is a ubiquitous issue in the field of modern molecular systematics. The widespread evolutionary discrediting phenomenon of ILS has been reported across the spectrum of life and cannot be simply explained in evolutionary terms as merely the remnants of ancestral polymorphisms. This is especially true when ILS occurs in mosaic patterns that defy common ancestry in a particular lineage and cannot be explained through sexual transfer.
Not only do homologous gene sequences produce ILS disparities in phylogenies, but so does the miRNA gene sequences that regulate virtually all forms of gene expression in the cell. When used in phylogenetic trees, miRNAs produce phylogenies that commonly contradict the inferred evolutionary trees as well as those developed with protein coding gene sequences. In fact, miRNAs often appear suddenly in taxa with no evolutionary precursors which particularly clouds interpretations of macro-evolution given that miRNAs are integrated into the cells’ genetic network of regulation and appear to tolerate very little mutation.
Another key type of ‘rogue’ evolutionary data is provided by the ubiquitous presence of taxonomically restricted gene sequences (TRGs). These appear suddenly in taxon and have no sequence homology to genes in other organisms. In all sequenced genomes to date TRGs comprise approximately 10–20% or more of the genes identified. Their sudden appearance, functional complexity, and integration in the complex network of the genome have no evolutionary explanation.
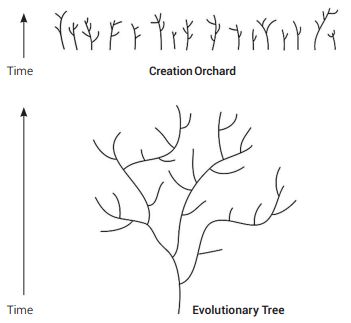
While commonalities across the spectrum of life can be observed in many gene sequences, this is a common theme inherent to engineered systems whereby similar mechanisms along with their control sequences show similar design. However, life is a mosaic of patterns as revealed in the many new genome sequences being produced and is not supportive of universal common ancestry, but rather the distinct creation of separate kinds of life as depicted in the Genesis account of origins. These separate kinds then diversified horizontally to produce what has been termed a ‘Creationist Orchard’ in contrast to the typical depiction of the standard tree of life (see figure 5).59
Creationists maintain that the original ‘created kinds’ have diversified (horizontally) over time and through such genetic bottlenecks as the global flood. Thus, the mosaic of life observed in DNA sequence fits well with this model. In 2006, Todd Wood published a comprehensive summary of the status of this concept within the creationist community.60
Perhaps DNA sequence data related to defining taxon (miRNAs and TRGs) that discredits evolution, alternatively could also be used on behalf of defining the genetic boundaries of created kinds.
References and notes
- Darwin, C., On the Origin of Species by Means of Natural Selection, or The Preservation of Favored Races in the Struggle for Life, 1859. Return to text.
- Zuckerkandl, E. and Pauling, L.; in: Bryson V. and Vogel, H. (Eds.), Evolving Gene and Proteins, Academic Press, New York, pp. 97–166, 1965. Return to text.
- Holder, M. and Lewis, P.O., Phylogeny estimation: traditional and Bayesian approaches, Nature reviews Genetics 4:275–284, 2003 | DOI:10.1038/nrg1044. Return to text.
- Throckmorton, L.H., Similarity versus relationship in Drosophila, Systematic Zoology 14(3):221–236, 1965. Return to text.
- Farris, J.S., Inferring phylogenetic trees from chromosome inversion data, Systematic Zoology 27(3):275–284, 1978 | DOI: 10.2307/2412879. Return to text.
- Dambroski, H.R. and Feder, J.L., Host plant and latitude-related diapause variation in Rhagoletis pomonella: a test for multifaceted life history adaptation on different stages of diapause development, J. Evolutionary Biology 20(6):2101–2112, 2007. Return to text.
- Koblmuller, S., Egger, B., Sturmbauer, C. and Sefc, K M., Rapid radiation, ancient incomplete lineage sorting and ancient hybridization in the endemic Lake Tanganyika cichlid tribe Tropheini, Molecular Phylogenetics and Evolution 55(1):318–334, 2010 | DOI: 10.1016/j.ympev.2009.09.032. Return to text.
- Elmer, K.R., Kusche, H., Lehtonen, T.K. and Meyer, A., Local variation and parallel evolution: morphological and genetic diversity across a species complex of neotropical crater lake cichlid fishes, Philosophical transactions of the Royal Society of London Series B, Biological Sciences 365(1547):1763–1782, 2010 | DOI: 10.1098/rstb.2009.0271. Return to text.
- Felsenstein, J., Alternative methods of phylogenetic inference and their interrelationship, Systematic Zoology 28(1):49–62, 1979 | DOI: 10.1093/sysbio/28.1.49. Return to text.
- Maddison, W.P. and Knowles, L., Inferring phylogeny despite incomplete lineage sorting, Systematic Zoology 55(1):21–30, 2006 | DOI: 10.1080/10635150500354928. Return to text.
- McCormack, J.E., Huang, H. and Knowles, L., Maximum likelihood estimates of species trees: how accuracy of phylogenetic inference depends upon the divergence history and sampling design, Systematic Zoology 58(5):501–508, 2009 | DOI: 10.1093/sysbio/syp045. Return to text.
- Boussau, B. et al., Genome-scale coestimation of species and gene trees, Genome Research 23(2):323–330, 2013 | DOI: 10.1101/gr.141978.112. Return to text.
- Degnan, J.H. and Rosenberg, N.A., Gene tree discordance, phylogenetic inference and the multispecies coalescent, Trends in Ecology and Evolution 24(6):332–340, 2009 | DOI: http://dx.doi.org/10.1016/j.tree.2009.01.009. Return to text.
- Churakov, G. et al., Mosaic retroposon insertion patterns in placental mammals, Genome Research 19(5):868–875, 2009 | DOI: 10.1101/gr.090647.108. Return to text.
- Grehan, J R. and Schwartz, J.H., Evolution of the second orangutan: phylogeny and biogeography of hominid origins, J. Biogeography 36(10):1823–1844, 2009 | DOI: 10.1111/j.1365-2699.2009.02141.x. Return to text.
- Ebersberger, I. et al., Mapping human genetic ancestry, Molecular Biology and Evolution 24(10):2266–2276, 2007| DOI: 10.1093/molbev/msm156. Return to text.
- Scally, A. et al., Insights into hominid evolution from the gorilla genome sequence, Nature 483(7388):169–175, 2012 | DOI: 10.1038/nature10842. Return to text.
- Felsenstein, J., Evolutionary trees from DNA sequences: a maximum likelihood approach, J. Molecular Evolution 17(6):368–376, 1981| DOI: 10.1007/BF01734359. Return to text.
- Kingman, J.F., Origins of the coalescent. 1974–1982., Genetics 156(4):1461–1463, 2000. Return to text.
- Wilkinson, M., Majority-rule reduced consensus trees and their use in bootstrapping, Molecular Biology and Evolution 13(3):437–444, 1996. Return to text.
- Clarke, G.D.P., Beiko, R.G., Ragan, M.A. and Charlebois, R.L., Inferring genome trees by using a filter to eliminate phylogenetically discordant sequences and a distance matrix based on mean normalized BLASTP scores, J. Bacteriol 184(8):2072–2080, 2002. Return to text.
- Talavera, G. and Castresana, J., Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments, Systematic Biology 56(4):564–577, 2007. Return to text.
- Castresana, J., Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis, Molecular Biology and Evolution 17(4):540–552, 2000. Return to text.
- Aberer, A.J., Krompass, D. and Stamatakis, A., Pruning rogue taxa improves phylogenetic accuracy: an efficient algorithm and webservice, Systematic Biology 62(1):162–166, 2013 | DOI: 10.1093/sysbio/sys078. Return to text.
- Degnan, J.H. and Salter, L.A., Gene tree distributions under the coalescent process, Evolution: International J. Organic Evolution 59(1):24–37, 2005. Return to text.
- Yu, Y., Than, C., Degnan, J.H. and Nakhleh, L., Coalescent histories on phylogenetic networks and detection of hybridization despite incomplete lineage sorting, Systematic Biology 60(2):138–149, 2011 | DOI: 10.1093/sysbio/syq084. Return to text.
- Rokas, A., Williams, B.L., King, N. and Carroll, S.B., Genome-scale approaches to resolving incongruence in molecular phylogenies, Nature 425(6960):798-804, 2003. Return to text.
- Kuo, C.H., Wares, J.P. and Kissinger, J.C., The Apicomplexan whole-genome phylogeny: an analysis of incongruence among gene trees, Molecular Biology and Evolution 25(12):2689–2698, 2008 | DOI: 10.1093/molbev/msn213. Return to text.
- Heled, J. and Drummond, A.J., Bayesian inference of species trees from multilocus data, Molecular Biology and Evolution 27(3):570–580, 2010 | DOI: 10.1093/molbev/msp274. Return to text.
- Kubatko, L.S., Identifying hybridization events in the presence of coalescence via model selection, Systematic Biology 58(5):478–488, 2009 | DOI: 10.1093/sysbio/syp055. Return to text.
- Than, C. and Nakhleh, L., Species tree inference by minimizing deep coalescences, PLoS Comput Biol 5(9):e1000501, 2009 | DOI: 10.1371/journal.pcbi.1000501. Return to text.
- Maddison, W.P., Gene Trees in Species Trees, Systematic Biology 46(3):523–536, 1997| DOI: 10.1093/sysbio/46.3.523. Return to text.
- Ashkenazy, H. et al., FastML: a web server for probabilistic reconstruction of ancestral sequences, Nucleic Acids Research 40:W580–584, 2012. Return to text.
- Thornton, J.W., Need, E. and Crews, D., Resurrecting the ancestral steroid receptor: ancient origin of estrogen signaling, Science 301(5640):1714–1717, 2003. Return to text.
- Chang, B.S., Jonsson, K., Kazmi, M.A., Donoghue, M.J. and Sakmar, T.P., Recreating a functional ancestral archosaur visual pigment, Molecular Biology and Evolution 19(9):1483–1489, 2002. Return to text.
- Chindelevitch, L., Li, Z., Blais, E. and Blanchette, M., On the inference of parsimonious indel evolutionary scenarios, J. Bioinformatics and Computational Biology 4(3):721–744, 2006. Return to text.
- Dolgin, E., Rewriting evolution: Tiny molecules called microRNAs are tearing apart traditional ideas about the animal family tree, Nature 486(7404):460–462, 2012 | DOI: 10.1038/486460a. Return to text.
- Pisani, D., Feuda, R., Peterson, K.J. and Smith, A.B., Resolving phylogenetic signal from noise when divergence is rapid: a new look at the old problem of echinoderm class relationships, Molecular Phylogenetics and Evolution 62(1):27–34, 2012 | DOI: 10.1016/j.ympev.2011.08.028. Return to text.
- Guo, L., Yang, S., Zhao, Y., Wu, Q. and Chen, F., Dynamic evolution of mir-17-92 gene cluster and related miRNA gene families in vertebrates, Mol. Biol. Rep. 40(4):3147–3153, 2013 | DOI: 10.1007/s11033-012-2388-z. Return to text.
- Axtell, M.J., Westholm, J.O. and Lai, E.C., Vive la difference: biogenesis and evolution of microRNAs in plants and animals, Genome Biology 12(4):221, 2011 | DOI: 10.1186/gb-2011-12-4-221. Return to text.
- Salmena, L., Poliseno, L., Tay, Y., Kats, L. and Pandolfi, P.P., A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language?, Cell 146(3):353–358, 2011 | DOI: 10.1016/j.cell.2011.07.014. Return to text.
- Miura, P., Shenker, S., Andreu-Agullo, C., Westholm, J.O. and Lai, E.C., Widespread and extensive lengthening of 3’ UTRs in the mammalian brain, Genome Research 23(5):812–825, 2013 | DOI: 10.1101/gr.146886.112. Return to text.
- Heimberg, A.M., Sempere, L.F., Moy, V.N., Donoghue, P.C. and Peterson, K.J., MicroRNAs and the advent of vertebrate morphological complexity, Proceedings of the National Academy of Sciences of the United States of America 105(8):2946–2950, 2008 | DOI: 10.1073/pnas.0712259105. Return to text.
- Tarver, J.E., Donoghue, P.C.J. and Peterson, K.J., Do miRNAs have a deep evolutionary history? Bioessays 34(10):857–866, 2012 | DOI: 10.1002/bies.201200055. Return to text.
- Robinson, J.M. et al., The identification of microRNAs in calcisponges: independent evolution of microRNAs in basal metazoans, J. Experimental Zoology. Part B, Molecular and developmental evolution 320(2):84-93, 2013 | DOI: 10.1002/jez.b.22485. Return to text.
- Lyson, T.R. et al., MicroRNAs support a turtle + lizard clade, Biol.Lett. 8(1):104–107, 2012 | DOI: 10.1098/rsbl.2011.0477. Return to text.
- Meredith, R.W. et al., Impacts of the Cretaceous Terrestrial Revolution and KPg extinction on mammal diversification, Science 334(6055):521–524, 2011 | DOI: 10.1126/science.1211028. Return to text.
- Al-Shahrour, F. et al., Selection upon genome architecture: conservation of functional neighborhoods with changing genes, PLoS Comput. Biol. 6(10): e1000953, 2010 | DOI: 10.1371/journal.pcbi.1000953. Return to text.
- De, S., Teichmann, S.A. and Babu, M.M., The impact of genomic neighborhood on the evolution of human and chimpanzee transcriptome, Genome Research 19(5):785–794, 2009 | DOI: 10.1101/gr.086165.108. Return to text.
- Khalturin, K., Hemmrich, G., Fraune, S., Augustin, R. and Bosch, T.C., More than just orphans: are taxonomically-restricted genes important in evolution?, Trends in Genetics 25(9):404-13, 2009 | DOI: 10.1016/j.tig.2009.07.006. Return to text.
- Tautz, D. and Domazet-Loso, T., The evolutionary origin of orphan genes, Nature Reviews Genetics 12:692–702, 2011 | DOI: 10.1038/nrg3053. Return to text.
- Yang, L., Zou, M., Fu, B. and He, S., Genome-wide identification, characterization, and expression analysis of lineage-specific genes within zebrafish, BMC Genomics 14:65, 2013 | DOI: 10.1186/1471-2164-14-318. Return to text.
- Colbourne, J.K. et al., The ecoresponsive genome of Daphnia pulex, Science 331(6017):555–561, 2011 | DOI: 10.1126/science.1197761. Return to text.
- Jacob, F., Evolution and tinkering, Science 196(4295):1161–1166, New York, 1977 | DOI: 10.1126/science.860134. Return to text.
- Meyer, S.C., Signature in the cell: DNA and the Evidence for Intelligent Design, 1st edn, HarperOne, 2009. Return to text.
- Zhaxybayeva, O. and Doolittle, W.F., Lateral gene transfer, Current Biology 21(7):R242–246, 2011 | DOI: http://dx.doi.org/10.1016/j.cub.2011.01.045. Return to text.
- Dunning Hotopp, J.C. et al., Widespread lateral gene transfer from intracellular bacteria to multicellular eukaryotes, Science 317(5845):1753–1756, 2007 | DOI: 10.1126/science.1142490. Return to text.
- Boschetti, C. et al., Biochemical diversification through foreign gene expression in bdelloid rotifers, PLoS Genetics 8:e1003035, 2012 | DOI: 10.1371/journal.pgen.1003035. Return to text.
- Wise, K., Baraminology: a young-earth creation biosystematic method; in: Walsh, R.E. and Brooks, C.L. (Eds.), Second International Conference on Creationism, Creation Science Fellowship, vol. 2, Pittsburgh, PA, pp.345–360, 1990. Return to text.
- Wood, T.C., The Current Status of Baraminology, Creation Research Society Quarterly 43(3):149–158, 2006. Return to text.







Readers’ comments
Comments are automatically closed 14 days after publication.