

Journal of Creation 34(2):111–116, August 2020
Browse our latest digital issue Subscribe
The hemizygosity hypothesis—a novel genetic paradigm for baranomes

Heterostyly is a fascinating feature of the flowers of a variety of plant families. It both helps to promote sexual reproduction and prevents inbreeding. In the most common form of heterostyly, plants develop either flowers with a short style and long stamens or with a long style and short stamens. The fact that in nature only plants with either of both phenotypes are observed suggests that only one gene is involved. However, a peculiarity is that several clearly defined characteristics (style length, stamens length, pollen size) are simultaneously expressed, indicating the presence of at least three genes. Recent molecular genetics demonstrates heterostyly is determined by a single genetic locus with five genes, which is inherited as a single, so-called ‘supergene’. Interestingly, only one of the parental chromosomes delivers the supergene to the offspring, thus the trait is present in a hemizygous state. This observation can be taken as a novel genetic paradigm to understand how potential, useful traits could be cryptically present in baranomes, i.e. the undifferentiated and uncommitted genomes of created kinds.
Flowering plants have complex reproductive structures. The pollen-producing male part is called the stamen and is usually composed of a filament and an anther. The filament, when present, holds up the anther, which produces pollen. The pollen-receiving female part is called the pistil. This is usually divided into three sections called the stigma, style, and ovary. Upon close examination, the flowers of primroses appear as two distinctly different types. Some flowers have long styles and short stamens; others have short styles and long stamens. The combination of a long style with short stamens is called the L-(long) phenotype, whereas the combination of a short style with long stamens is called the S-(short) phenotype (figure 1). In botany, this dimorphism is called heterostyly—a phenomenon described by Charles Darwin. In 1860, Darwin wrote a series of letters to his friend and confidant J.D. Hooker and to the botanist J.S. Henslow. He had studied two primrose species and reported that these consistently produced two types of flowers. Both species existed in two forms of approximately equal numbers, which appeared to be ‘male’ and ‘female’. The stamens in the supposed ‘female’ form had very short filaments. It also produced small elongated pollen grains, and the pistil had a long style with long stigma papillae. In the supposedly ‘male’ form, he reported “long filaments, large round pollen grains, and a short pistil with short papillae”.

Darwin recognized that the different types of flowers represented different reproductive systems, which he called ‘distyly’. He correctly argued that both flower types were hermaphrodites and that “the pollen of A is adapted to the stylus of B, and vice versa”. In the following years, Darwin published his observations on heterostyly, especially its occurrence in flax (Linum) and willow (Lythrum). In 1877, he compiled his research on heterostyly in the book Different Forms of Flowers. As a result of this work, Darwin is widely recognized as the first to study heterostyly in primroses and as the one who provided the functional significance of the two types of flowers.1
Species expressing heterostyly often have so-called plate-like flowers, whose lower part is tubular in shape. The stamens are fused with the flower tube. In the L-phenotype, this leads to pollen compartments in the flower tube being fixed in a low position. In the K-phenotype flowers, the pollen compartments are positioned much higher, towards the opening of the flower tube.2,3 It is easy to imagine the purpose of this arrangement: flower-visiting insects transport the pollen from the short stamens to flowers with short pistils, and vice versa, due to the places where the respective pollen sticks to an insect’s body. It is a very clever design to promote and maintain sexual reproduction. Yet, the genetics of the situation are complex and surprising. The history of how we figured out the biology and genetics of heterostyly is also very interesting.
Genetics of heterostyly
Most experimental crosses with primroses suggested that the traits of the L- and K-flowers were inherited as if it were due to a single allele pair (S and s). The site is now called the S-locus (a ‘locus’ is a specific site on a chromosome). Primroses of the K-phenotype are therefore S/S homozygotes and S/s heterozygotes, while those of the L-phenotype can be understood as s/s homozygotes. Nevertheless, so-called homostyle flowers in which the style and the stamens are on the same level also exist (figure 1). These flowers show that the characteristics are determined by at least two separate but closely linked genes.4 This was known for quite some time, but then some very rare and unusual flower types were found, some of which had long stamens and small pollen grains, others short stamens and large pollen grains. They proved that pollen size can be inherited independently and so an extra gene had to be postulated. The new types of flowers had to be caused by new alleles in the S-locus, which led to the conclusion that it represents at least three very closely linked genes, which determine the different characteristics of heterostyly.5
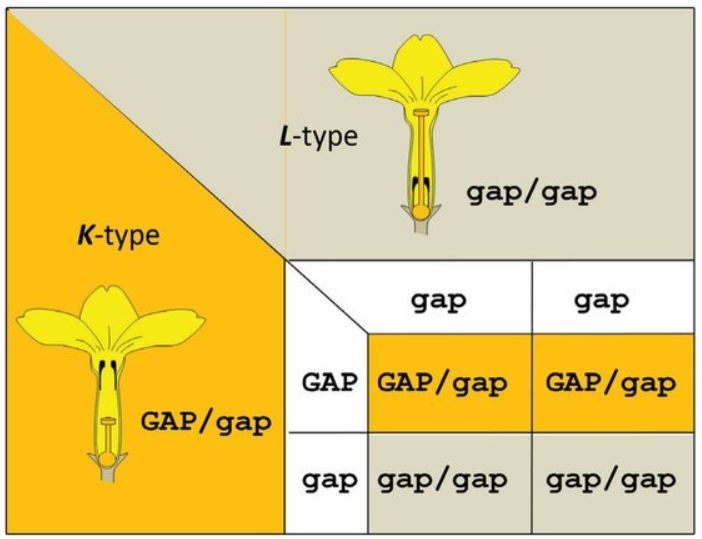
Note that thus far in the discussion, a ‘gene’ is being defined as ‘an inheritable unit’. All this work was done before we knew what the units of inheritance were (i.e. stretches of DNA). Thus, at first, we knew that two flower types existed, so this was put down as the result of two different forms of the same ‘gene’. We then realized that additional flower types also existed, so the first ‘gene’ had to be subdivided into several more. Following Mendelian genetics, these genes should have dominant and recessive alleles. In this view, the first gene (with alleles G and g) would determine the length of the style and papillae, the second gene (with alleles A and a) would determine the length of the stamens, while the third gene (with alleles P and p) would determine pollen size and thus male compatibility. Since all three genes are usually associated with each other, the K-phenotype thus has the genotype GAP/gap and the L-phenotype gap/gap. Crosses between GAP/gap and gap/gap plants always produce K and L phenotypes in a 1:1 ratio (figure 2), which corresponds to observations in nature. This view prevailed until the 1990s. Why GAP/GAP did not exist in nature was unknown. The extremely rare homostyle flowers were thought to be due to a recombination event of the chromosomes, in which the genes are exchanged, and in this way the atypical combination formed. It was also suspected that two other genes should exist responsible for female and male incompatibility, since it would be unlikely that a single gene could control both morphological and incompatibility aspects of the flowers.6 Thus, the S-locus appeared to be a so-called supergene, a chromosomal segment with several tightly linked individual genes that together control an integrated phenotype comprised of several traits.
Supergenes are always inherited as a single group and stand as an excellent genetic design principle, which enables programmed adaptations that can be released in only one or a few steps. Recently, supergenes have been found to control mimicry in butterflies and sexual dimorphism in ruffs (the bird Philomachus pugnax), as well as adaptations in honeybees and zebrafish.7 In primroses, the supergene may not only explain heterostyly, but also the occurrence of very rare homostyle flowers and pollen incompatibility. The identity and function of the genes present in the S-locus has only recently been unravelled, as well as a rather surprising explanation for the dominant and recessive alleles.
Molecular genetic analysis of the S-locus
In the 20th century, genetic studies on primroses made it possible to create a genetic map of the S- locus. Furthermore, sequencing the genome of Primula veris (the cowslip, or cowslip primrose) led to a reference genome sequence from a pool of L- and K-phenotypes. Subsequent transcriptome analyses identified the genes that were most strongly expressed in both flower types.8,9 These studies formed the basis for two important breakthroughs in our understanding of heterostyly in primroses: the identification and functional description of the complete S-locus in the L- and K-phenotypes.10,11 They showed that the S-locus is located right next to the centromere of the largest chromosome, which would explain why hardly any recombination occurs in the S-locus. When geneticists speak of recombination, they mean the exchange of alleles present on homologous chromosome arms (figure 3). If a gene is located near the centromere, recombination hardly occurs, and genetic exchange is rare. As a result, the inheritance of the entire complex of traits behaves like a single Mendelian locus (as if it were only one trait) and this situation results in the L-and K-phenotypes remaining in approximately equal amounts.

Only recently have genes been identified that orchestrate heterostyly. In 2016, it was reported that the flowers of the K-phenotype express several unique genes, which were absent in the L-phenotype.9 As expected, genes for style length, stamen length, and pollen size were found in the dominant S-locus, as well as genes for female and male incompatibility. Of the five genes identified, two had been known previously: CYP734A50 and GLO2. The CYP734A50 gene codes for a cytochrome P450 enzyme that inactivates a class of cell growth hormones (the so-called ‘brassiosteroids’), which makes it understandable that a mutated CYP734A50 gene is associated with long homostyle flowers. The GLO2 gene belongs to a family of morphogenetic genes that control and regulate genetic programs that determine the plant’s shape and size. Indeed, an inactive GLO2 gene had been associated with short stamens. So, the CYP734A50 gene determines the style length and possibly the female incompatibility, whereas the GLO2 gene is responsible for the length of the stamens.6 Hence, these two genes form the basis for the G/g and A/a alleles of classical genetics. The remaining three genes code for a Kelch-repeat F Box protein (KFB), a pumilio-like RNA-binding protein (PUM) and for a protein with a highly conserved C-terminal domain (CCM). It is not yet fully understood how these genes are related to phenotype, although KFB and PUM proteins seem to be responsible for pollen size and male incompatibility.6 In addition, a so-called CFB gene was found flanking both sides of the dominant S-locus (see figure 2).The most fascinating observation was, however, that the recessive s-locus was about 280,000 DNA letters shorter than the dominant S-locus. The s-locus contained only a single CFB gene and there was no trace of the other genes.6 The S-locus in primroses is therefore hemizygous, i.e. a unique DNA region that occurs only once (mono-allelic) per genome. This finding provides an elegant explanation for the suppressed recombination found in earlier studies, since there simply is no corresponding counterpart for the exchange of genes in the s-locus. These recessive traits are not caused by genes, but by the absence of genes!
The hemizygosity hypothesis
Previously, the term ‘baranome’ was introduced to describe the genetics of the created kinds.12 This concept was introduced because the term ‘genome’, as it is commonly used in genetics, is not sufficient to describe the genetics of created kinds. To explain the rapid diversification of life after the Flood, biblical creation requires created kinds to be geared with pluripotent, undifferentiated, and uncommitted genetic information carriers with an intrinsic ability for rapid adaptation and speciation. Thus, created kinds must have cryptic genetic information contained within them. They must have been hardwired with programs to bring forth novel species (after their kind) and mechanisms to induce variation and adaptations. Over the past two decades several of these programs have been discovered: epigenetics13 and transposable and transposed elements (TEs).14,15 In addition, there are several ways to program extra information or variation into the chromosomes of a created kind. As argued by Carter, most of the common allelic variation we observe in humans today may have been ‘created diversity’ and would have been present on the original chromosomes in Eden.16 This suggests that other created kinds contained many heterozygous genes (‘alleles’). Furthermore, it has been argued that baranomes may have been preloaded with mechanisms designed for the rapid induction and/or release of cryptic information by TEs.14,15 It should be noted that in the creation paradigm TEs could be renamed ‘variation-inducing genetic elements’ (VIGEs).17 There may be more characteristics of modern genomes that indicate what baranomes may have originally looked like. The hemizygous S-locus with five unique genes in primroses provides an additional paradigm for preloaded cryptic genetic information. Created kinds may have contained many features that were originally hemizygous, which could easily become fixed or lost through population dynamics (i.e. selection, genetic drift, etc.). With preloaded hemizygous genetic information, adaptation and formation of new species does not necessarily require millions of years. It may, in fact, occur in the blink of an eye. Due to population dynamics and VIGE activity, preloaded hemizygous information may also rapidly disappear from differentiating baranomes/genomes.
Hemizygous variation within species is huge
Until recently, it was tacitly assumed that all individuals within a single species should contain approximately the same number of genes. This assumption, however, has proven to be wrong. Recent molecular genetics demonstrated that the genomes of organisms belonging to the same species are so variable that geneticists today rather assume that there is no such thing as ‘a species genome’. For example, in the wild forms of the mouse-ear cress (Arabidopsis thaliana), approximately 4% of the genes are completely different from the reference genome or are not found at all.18 Something very similar can be observed in the K- and L-phenotypes of primroses. A comparison of the genomes of the two phenotypes would show that at least five genes are missing in the L- plants: GLO2, CYP734A50, KFB, PUM, and CCM. The strict separation of these hemizygous genes on different chromosomes is best interpreted as evidence of programmed variation, especially since there is no plausible evolutionary explanation for the existence and multiple independent occurrence of these genes: heterostyly has emerged independently at least 20 times in different 28 plant families.2 Even within families, such as the Boraginaceae (forget-me-nots and borages), several independent origins have been reported.19 Rather than independent acquisition, the Boraginaceae may have repeatedly lost the genes responsible for heterostyly, so that the trait was maintained in a hemizygous way in several members of this family and lost in others. Moreover, in the six genera studied so far, heterostyly also appears to be controlled by a single locus that is very similar to the supergene of primroses.20 Explained away as ‘convergent evolution’ by our Darwinian friends,21 an intelligent design signal could not be stronger.

The Human Genome Project and the 1,000 Genomes Project lend credence to the ‘hemizygosity hypothesis’. Structural variations—such as insertions and deletions, but also inversions and translocations—are very important genetic variants, but until recently our techniques to identify them lagged behind those developed to identify single nucleotide polymorphisms (SNPs). The human reference genome was initially built with approximately 2/3 of the data coming from one single individual genome and on false ideas that human genetic diversity was rather limited. As more genomes were sequenced, it became more obvious that individual genomes are much more variable than originally anticipated. The sequencing and de novo assembly of a Korean individual filled more than 100 gaps in the reference genome.22 Since 360 megabases (12%) of the genomes of 270 individuals with ancestry in Europe, Africa, and Asia differ due to indels (insertions and deletions, which may be part of created diversity or due to de novo mutation), every individual has a unique pattern of gains and losses of complete sections of DNA.23 Imagine what happens if a Korean and an African individual would marry and produce offspring. Indeed, a significant part (several percent) of the DNA sequences of the genomes of the children might come in a hemizygous state! Numerous child-rich marriages between individuals of different human populations are evidence that hemizygous genomes easily match and lead to fertile offspring. This shows that hemizygosity does not interfere with reproductive success.
The hemizygosity hypothesis may also explain why we find a high number of indel-differences in different Homo subspecies (i.e. Neandertal, Denisovan, and modern man). In 2011, 584 human-specific conserved indels (short: hCONDEL) were found and examined in more detail. These sequences with a median size of 2,804 bp are found in chimpanzees but seem to be completely missing in the human genome. They are found almost exclusively in non-coding regions, often in the vicinity of genes involved in steroid hormone signal transduction and neuronal function, where they can serve as regulatory elements for gene expression. They are claimed to be tissue-specific enhancers that may underlie the regulatory changes necessary for the evolutionary divergence of humans and chimpanzees.24 Interestingly, about half of these hCONDELs were detected in archaic humans (Neandertals and Denisovans), indicating that they have disappeared from the genome of modern humans.25 Mendelian genetics and genetic drift are able to readily reduce hemizygosity to homozygosity in small and isolated populations. Therefore, hemizygosity may explain why we observe DNA sequences in Neandertals that are completely missing from the modern human genomes. It should be noted that sequences present hemizygously in our human ancestors did not only provide instant dominant/ recessive trait pairs; they could also be readily lost when such traits were not subject to strong selection. The disappearance of hemizygous regions from the genome is not very hard to conceive since all studied genomes contain elaborate genetic engineering mechanisms in the form of VIGEs. VIGEs have the ability to restructure and recombine the genetic material instantly.12,13,26 Their omnipresence in the genome makes them excellent tools to understand the indel-mutations observed in the human subpopulations.23
Conclusion
It is essential to have a working creation science model that systematically and reliably explains variation, adaptation, and speciation in a biblical timescale. Many incisive researchers who take the Bible seriously have been working on such models for decades.
These efforts have met with limited success, mainly because the genetic basis for created kinds has long been elusive. Although preloaded genetic evolution models have been put forward for long timeframes,27 it is only recently that we begin to understand how it might work for short timeframes. In the 21st century we have come to understand the genome as a highly dynamic information storage and processing device that is prepared to adapt to changing environments. Organisms do not necessarily have to wait for random mutations to adapt and speciate, since their genomes contain preloaded programs and mechanisms that respond to all sorts of unexpected challenges. These super-sophisticated genomes have all the hallmarks of intelligent design and this is exactly what we would expect from a biblical perspective.
In this paper, a novel mechanism for rapid baranomic diversification was introduced and discussed using the hemizygosity concept of heterostyly in primroses. In diploid genomes, significantly more information can be stored than previously thought possible if some of that information is stored hemizygously. Due to genetic recombination and Mendel’s laws of heredity, the unique hemizygous information can then simply be spread across different populations (figure 4). Isn’t there grandeur to this view of life, where God chose to create life as adaptive, free-living entities equipped with pluripotent baranomes, so that the earth could bring forth living creatures—livestock and creeping things and the beasts of the earth—according to their kinds?
References and notes
- Gilmartin, P.M. On the origins of observations of heterostyly in Primula, New Phytol. 208:39–51, 2015. Return to text.
- Ganders, F.R. The biology of heterostyly, New Zealand J. Bot. 17:607–635, 1979. Return to text.
- Charlesworth, D. and Charlesworth, B., A model for the evolution of distyly, Amer. Nat. 114:467–498, 1979. Return to text.
- Danish botanist Wilhelm Johannsen coined the word ‘gene’ (‘gen’ in Danish and German) in 1909 to describe these fundamental physical and functional units of heredity. The terms gene and genetics (introduced by geneticist Bateson in 1906) were coined long before Watson and Crick discovered that the DNA was a double helix of nucleotide sequences. Return to text.
- Ernst, A., Heterostylie-Forschung: Versuche zur genetischen Analyse eines Organisations- und ‘Anpassungs’ Merkmales, Z. Ind. Abst. Vererb. Lehre 71:156–230, 1936. Return to text.
- Kappel, C., Huu, C.N., and Lenhard, M.A., Short story gets longer: recent insights into the molecular basis of heterostyly, J. Exp. Bot. 68:5719–5730, 2017. Return to text.
- Pennisi E., ‘Supergenes’ drive evolution, Science 357:1083, 2017. Return to text.
- Nowak, M.D., Russo, G., Schlapbach, R., Huu, C.N., Lenhard, M., and Conti, E., The draft genome of Primula veris yields insights into the molecular basis of heterostyly, Genome Biology, 16:12–19, 2015. Return to text.
- Huu, C.N., Kappel, C. et al., Presence versus absence of CYP734A50 underlies the style-length dimorphism in primroses, eLife 5:e17956, 2016. Return to text.
- Li, J., Cocker, J.M., Wright, J. et al., Genetic architecture and evolution of the S-locus supergene in Primula vulgaris, Nature Plants 2016 2:16188, 2016. Return to text.
- Burrows, B.A. and McCubbin, A.G., Sequencing the genomic regions flanking S-linked PvGLO sequences confirms the presence of two GLO loci, one of which lies adjacent to the style-length determinant gene CYP734A50, Plant Reproduction 30:53–67, 2017. Return to text.
- Terborg P., Evidence for the design of life—part 2: baranomes, J. Creation 22(3):68–76, 2008. Return to text.
- Ambler, M., Epigenetics—an epic challenge to evolution, creation.com/epigenetics-challenges-neo-darwinism, 2015. Return to text.
- Terborg, P., Evidence for the design of life—part 3: an introduction to variation-inducing genetic elements, J. Creation 23(1):99–106, 2009. Return to text.
- Terborg, P., Evidence for the design of life—part 4: variation-inducing genetic elements and their function, J. Creation 23(1):107–114, 2009. Return to text.
- Carter, R.W., Effective population sizes and the loss of diversity during the flood bottleneck, J. Creation 32(2):124–127, 2018. Return to text.
- I propose that we make our own creation paradigm jargon, here, since that of our opponents is often biased, and sometimes even misleading. Evolutionary concepts like ‘junk DNA’ and ‘pseudogenes’ turned out to be completely wrong and antiscientific. The term ‘non-coding DNA’, which is now used to replace ‘junk DNA’, is still a misnomer, since it is coding for biological function, usually regulatory RNA molecules. Return to text.
- Clark, R.M., Schweikert, G. et al., Common sequence polymorphisms shaping genetic diversity in Arabidopsis thaliana, Science 317:338–342, 2007. Return to text.
- Cohen, J.I., A phylogenetic analysis of morphological and molecular characters of Lithospermum L. (Boraginaceae) and related taxa: evolutionary relationships and character evolution, Cladistics 27:559–580, 2011. Return to text.
- Lewis, D. and Jones, D.A., The genetics of heterostyly; in: Barrett, S.C.H. (Ed.), Evolution and Function of Heterostyly, Springer, Berlin, Germany, pp. 129–150, 1992. Return to text.
- Zhong, L., Barrett, S.C.H. et al., Phylogenomic analysis reveals multiple evolutionary origins of selfing from outcrossing in a lineage of heterostylous plants, New Phytol. 224:1290–1303, 2019. Return to text.
- Lee, C., Greater diversity is needed in human genomic data, The Scientist 9: 26–27, 2019. Return to text.
- Redon, R., et al., Global variation in copy number in the human genome, Nature 444:444–454, 2006. Return to text.
- McLean, C.Y., Reno, P.L. et al., Human-specific loss of regulatory DNA and the evolution of human-specific traits, Nature 471:216–219, 2011. Return to text.
- Cserhati, M.F., Mooter, M.E., et al., Motifome comparison between modern human, Neanderthal and Denisovan, BMC Genomics 19:472–483, 2018. Return to text.
- VIGEs are variation-inducing genetic elements. I defined them in my first VIGE paper in 2008 as all sorts of repetitive sequences, including TEs. Their repetitive character makes them excellent tools for recombination events. There is even a VIGE (a DNA transposon), which holds the genetic info for recombination enzymes. Further, a high number of recombination events are initiated from well-determined methylation events on TEs. This shows that recombination is not a random process, but is regulated and controlled. Return to text.
- Davison, J.A., An evolutionary manifesto: a new hypothesis for organic change, hedgeschool.com/Essays_pdf_Home/06_Science_PDF/06_Science_Essay_Evolution_Davison_Manifesto_Evolutionary.pdf, 2000. Return to text.




Readers’ comments
Comments are automatically closed 14 days after publication.