

Journal of Creation 35(2):98–106, August 2021
Browse our latest digital issue Subscribe
The surprisingly complex tRNA subsystem: part 5—evolutionary implausibility
The genetic code is contingent on several indispensable components. Parts 1–4 of this series documented how dozens of protein molecular machines are necessary to prepare and process the members of one subsystem, the collection of tRNAs. But without functional linker molecules the necessary protein cannot be manufactured. Evolution theory is incompetent to solve this chicken-and-egg problem.
Given a putative, primitive ancestral code, mathematical modelling demonstrates that at most four precise mutations (or the statistical equivalent in the case of alternative solutions) could have been generated in 10 billion years. This does not provide a feasible means to produce various features, including: a) the multiple tRNA-specific enzymes involved in some chemical modifications; b) the precise 7-nt target sites found in several Drosophila genomes perfectly complementary to the regulating 5’-tsRNAs; nor c) miRNAs derived from tRNAs having perfect seed regions of about 7 nucleotides.

In part 1 of this series, we showed the genetic system is composed of several indispensable subsystems.1 Cellular subsystems consist of multiple biochemicals assembled in a precise time order with elaborate quality control measures. They function holistically, involve several processing steps, and are integrated with other cellular subsystems. To illustrate the concept of a subsystem, consider the eukaryote intron splicing cycle.2-4 A spliceosome assembles and then disassembles nine discrete complexes sequentially, using different ensembles of snRNAs (small nuclear RNAs) and proteins. Some of the biomolecules are only needed temporarily to produce the next complex in the pathway. Maturation of many of the sub-assemblies requires trafficking between the nucleus and cytoplasm for special processing, with many quality-control measures to prevent errors. Over 200 proteins are involved, and all the complexes consume much energy, but only the last complex provides any value. Understanding in detail the design principles of one subsystem provides key insights to understand the hundreds of other subsystems found in cells (such as miRNA processing, nuclear pore trafficking, horizontal gene transfer, and so on).
Various evolutionary theories postulate either a DNA, RNA, or protein-only predecessor to extant life. A theory requiring multiple classes of pure, complex biomolecules at the right time and place would not be consistent with naturalism. However, deeper analysis of genetic subsystems reveals they are all dependent on DNA, RNA, and proteins concurrently to be viable. The subsystems themselves consist of parts which are irreducibly complex.
The motivation for this five-part series was to analyze the genetic system, but focusing for now on the simplest subsystem, a collection of transfer-RNAs (tRNAs).1 Many assume only RNA molecules would be necessary to serve as adaptor molecules to translate codons. Knowing the other subsystems are considerably more complex, such as ribosomes5 and aminoacyl-tRNA-synthetases,6-9 we chose to defer analyzing them for later.
The protein-based synthetases are responsible for attaching the correct amino acid in a high-energy bond to the correct tRNA at a precise nucleotide (nt) position. What came first? Without aminoacyl-tRNA-synthetases proteins will not be produced, but aminoacyl-tRNA-synthetases consist of proteins. Without aminoacyl-tRNA-synthetases, tRNAs are worthless as translation adaptor molecules. Without tRNAs, aminoacyl-tRNA-synthetases are worthless. Crucially, a living cell cannot exist without a functioning genetic system, comprising all its interacting subsystems (see part 1).
Dozens of kinds of tRNAs are needed to translate the genetic code, codon by codon to produce the protein sequences specified on DNA. In part 1, we showed that transcribing and processing pre-tRNAs in the appropriate amount and right time requires protein-based molecular machines in all life-forms.1 In parts 2–4 we provided insights on how tRNAs are processed, trafficking between the nucleus and cytoplasm multiple times.10-12 We also described many cellular processes they are involved in.
Here in part 5 we will consider whether evolution is a plausible explanation for these observations. Evolution faces two conceptual problems. The first is the absence of a simple but realistic starting point for the tRNA subsystem. Second, should a minimally viable primitive genetic system have arisen naturally, could the additional processes in which tRNAs are involved in have arisen through evolutionary processes? Before we examine both questions, we must be cognizant of some key principles.
Minimal functionality for a new feature to arise

Entirely new biological features are not built with only one nucleotide. Many nucleotides must participate jointly, as an entity which serves as a starting point with at least minimal functionality (see the red box in figure 1).
To illustrate, suppose a trinucleotide ‘ATG’ is to serve as the Start codon for translation at a genome position which currently contains CCC. Intermediate mutations such as CCC → ACC → ATC → are not advantageous whenever one more nucleotide matches the functional pattern. In this example, the single-nucleotide mutational path would require no less than three single-nucleotide mutations at specific locations before any benefit accrues. But why should a ‘correct’ mutation always occur? A more convoluted path would be expected, such as CCC → GCC → GTC → GTT → ATT → ATA → ATG.13
Multiple mutations destroy features more rapidly
Perhaps a few fortuitous double mutations could occur in a mutation-prone region. Might this facilitate obtaining the right pattern, through a sequence of mutations such as CCC → ATC → ATG? It turns out that multiple concurrent changes would accelerate destruction of the pattern just generated. Let us elaborate.
Perhaps the pattern needed is within one or more hot mutational spots or recombination areas. The example in figure 2 proposes that the ATG pattern is located within a 6 nt high mutation region. We see that 3/6 of the possible point mutations would destroy the function; 12/15 of the double mutations; and 18/19 of the triple mutations would do so. (The rapidly mutating region need not be adjacent; the pattern could be distributed across multiple such regions). We conclude that random mutations will destroy a specific, useful pattern faster when: it is comprised of more nucleotides; mutation rates in that region are higher; and multiple concurrent mutations occur.

In the analysis below we require only minimal functionality, to facilitate a theoretical evolutionary starting point. Consequently, selective advantage would be small at such time, precluding rapid spread through a population. The mutated members would be highly vulnerable to destruction by additional mutations.
‘Statistic equivalency’ to analyze necessary mutations
In the example above, the ATG pattern consists of adjacent nucleotides which are intolerant to mutations. This is the simplest case to analyze mathematically. More complex examples would require using Bayesian statistics or Shannon information theory. However, our purpose here is to evaluate whether evolution is a reasonable explanation for the tRNA subsystem, and we wish to avoid unnecessary side-tracking complexity.
There are many examples of the conceptually simplest case which involve invariant adjacent nucleotide patterns, such as the seed location of miRNAs (red nucleotides in figure 3A). Various binding sites on DNA, RNA and proteins have this property and are generally described using consensus sequences or sequence logos.14
Scenario B of figure 3 shows how requiring either of two nucleotides at two precise locations is statistically equivalent to requiring a certain nucleotide in one position. In some cases, multiple separated patterns are needed (scenario C in figure 3). An example is the individual patterns necessary to recognize the 5’ and 3’ ends of an intron plus the branch point.15
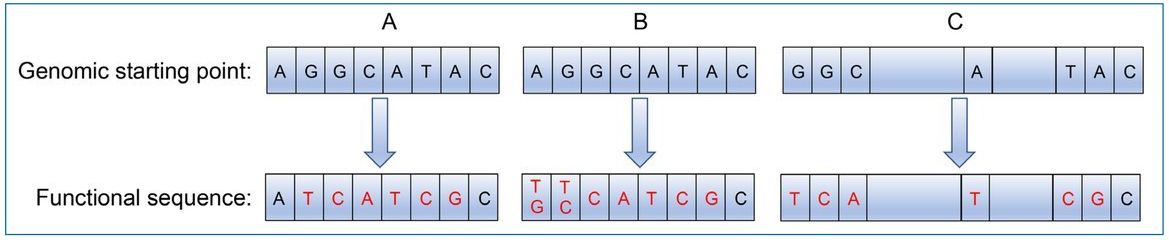
Additional informational patterns outside of key highly invariant positions are usually necessary to ensure correct targeting. Identifying and characterizing the additional requirements can be difficult, which is why bioinformatics algorithms are less than 100% accurate.16,17 We conclude that the necessary informational patterns can be a combination of invariant positions along with a subset of acceptable nucleotides at other positions.
We now introduce the notion of ‘statistical equivalency’. In some cases, a specific nucleotide might not be necessary at a genomic position. Instead, a genetic solution could be satisfied through multiple collaborating nucleotides. This makes design sense, permitting rheostat-type activity levels. Many alternative patterns may work, but with different durations or rates.
Obtaining multiple but less stringent mutations by chance is often less probable than requiring a single specific mutation. This can be observed from sequence logos which permit probability calculations. A portion of a pattern containing more than two bits of Shannon information is less probable than requiring a specific nucleotide out of four possibilities, since log2(4) = 2. Therefore, the pattern in figure 3B can be considered to contain the equivalent of at least six invariant nucleotides, especially when one knows that additional unidentified signals are necessary.
Maximum number of specific mutational events
In The Edge of Evolution, Behe estimated the maximum number of specific mutations which could be demanded from an evolutionary explanation as the starting point for an innovation.18 He used empirical examples which required one or two specific amino acids be modified.
P. falciparum, a eukaryotic parasite transmitted by mosquitoes, is the main cause of malaria. Various medications have been used to kill this parasite. A single point mutation in the cytB gene confers resistance to atovaquone and a single point mutation in the dhfr gene resistance to a different drug, pyrimethamine. Medical studies have established that 1012 parasite multiplications are necessary for each de novo case of parasite resistance to these two drugs.19
An adult with malaria carries ~1012 parasites, but in the laboratory much higher mutation rates than 10-12 were recorded.19 Why is parasite immunity not arising more frequently? One possibility could be that the mutations lead to fitness disadvantages until exposed to the drug. In addition, human immunity might prevent some of the resistance-conferring mutations from being effective.19
These are important considerations. Another fact is that many random mutations will be ‘wasted’ in the exploratory process, generating identical mutations and mutational reversals. If one out of n genomic positions must be mutated, it will take far more than n random mutations to ensure every position has been ‘tested’.
P. falciparum feeds on the protein part of hemoglobin but the heme part is poisonous. Heme is normally processed in the parasite into hemozoin which can be disposed of. The drug chloroquine functions by inserting itself into the parasite’s digestive vacuole where it interferes with the production of hemozoin. The protein transport pump PfCRT is responsible for importing various substances into the vacuole, fortuitously also the therapeutic chloroquine. Analysis of all P. falciparum lineages which are resistant to chloroquine revealed that the same two amino acids had mutated, at positions 76 and 220 of PfCRT.19,20
Medical studies show that less than 10 independent cases of spontaneous immunity have arisen since chloroquine was first used. It is also known that the malarial parasite genome consists of about 108 nucleotides. Taking into account the average number of infected worldwide per year and parasites per sick person led to the conclusion that about 1020 P. falciparum cells would need to mutate to confer a single case of immunity, which involved exactly two amino acids.19 Since most of the unsuitably mutated versions would be killed by the drug, the ‘correctly’ mutated would face fewer competitors in a cell and reproduce rapidly. Transmission by mosquitoes to new hosts would quickly spread the resistant strain, and therefore chloroquine is now usually ineffective.
When P. falciparum is no longer exposed to chloroquine the proportion of mutant strain declines and the original strain dominates. Although less fit in almost all environments the double mutation is indispensable for survival of the parasite in the presence of this drug.21
Since mutating a nucleotide in a codon in most cases will code for a different amino acid, we can continue the analysis by referring to nucleotides. We have established so far that empirically about 1012 mutational events would be necessary for a single cell eukaryote having a genome of about 108 nucleotides to achieve a specific evolutionary novelty, and 1020 mutational events when two specific mutations are needed.
Most of the organisms which have lived on earth are prokaryotes and about 1030 are produced worldwide annually.22 Since evolutionists believe life on earth began less than 10 billion years ago (1010) then less than 1040 mutations could have ever occurred.
Behe pointed out that if about 1020 organisms would be needed to produce two specific mutations, then 1040 would be necessary for an evolutionary innovation initiating from four mutations.18 The limitation to four or less specific mutations applies to every step of every putative evolutionary trajectory (figure 4A). This illustrates the power of Behe’s concept of irreducible complexity. If evolution is true, then complex innovations which require multiple new and interacting genes, such as to create the cilium, must have occurred though countless relatively small steps, all without a guiding plan.23

Realistic maximum number of specific mutational events
Far fewer than the maximum number of random mutational events, 1 × 1040, could be applied to develop a particular innovation for several reasons.
- Far more mutations are deleterious than beneficial. According to a review article written by two specialists, for Drosophila and enteric bacteria the proportion of deleterious amino-acid-changing mutations is at least 84% and 97.2%, respectively. Requiring several specific amino acids is therefore especially difficult.24 Comparison across many organisms has revealed far less variability than expected if non-protein coding sites were mutationally neutral. The ‘Junk DNA’ myth has been very damaging to science and has strongly influenced speculative claims about a high proportion of effectively neutral mutations. For organisms such as yeast, nematodes, and Drosophila melanogaster about 50% of all mutations may be deleterious.24
- In three mutagenesis experiments the proportion of advantageous mutations obtained were 0% in Escherichia coli, 0% in the bacteriophage Φ6, and 6% in Saccharomyces cerevisiae. Therefore, lineages accumulating more mutations will on average have proportionally fewer members. This makes it increasingly difficult to collect enough changes to generate a fortunate genetic pattern natural selection could act on.25
- The stochastic nature of survival and reproduction across many generations will cause most useful mutations to be lost over deep time instead of fixing in the population.26
- For fixation to occur, the cells with a beneficial mutation must out-compete all those lacking it, wiping out all alternative evolutionary possibilities (figure 5). This would happen again and again for every discrete level of improvement shown in figure 4A.
- The relevant mutations must occur within a specific class of organism.
- The mutations must occur within a relevant time window.
- The difference between standing (N) and the effective (Ne) population sizes must be considered. A recent study of 153 species (152 bacteria and one archaeon) led to estimated effective population sizes Ne on the order of 108–109, even though their census numbers are orders of magnitude higher.27 The cyanobacterium Prochlorococcus marinus is the most abundant photosynthetic organism on Earth. Census populations can be upwards of 1013, but the Ne is only about 108.

An excellent way to research this topic is by using Mendel’s Accountant, the leading population genetics simulation program available.28
The above analysis indicates that fewer than the statistical equivalent of four specific mutations can be invoked to claim a naturalistic origin for a cellular novelty. Innovation, however, is not the same of loss of function. If in a particular environment an organism no longer needs a complex feature, any mutation able to deactivate it will be beneficial. Material and energy would be saved along with fewer opportunities for genetic errors. Therefore, the opportunities to simplify and streamline will not be limited to just one mutation at a single genomic site.
Improbability as merely a statistical outlier?
Statistically unlikely events do occur. Perhaps we would be over-rating a fortunate evolutionary accident upon finding a four specific mutation example. To judge this possibility, one needs to examine several examples and in more detail. Obtaining a Start codon at the correct location to define where to begin translation in one case is remarkable. But finding this thousands of times in the same prokaryote species cannot be shrugged off as a curiosity, especially if we include additional requirements such as the need of a suitable termination codon. But more significantly, a Start codon alone is worthless without the new required proteins which interact with it and each other.
Let us review what is involved in a minimal protein-protein binding interaction. After folding into a stable three-dimensional shape, part of a protein surface must offer a series of indentations and projections which act like a lock and key. Shape alone is not enough; chemical electrostatic properties are also needed (using the right amino acids with net positive and negative charge distributions) as well as hydrophobic surface patches.
Dr Winter and co-workers at the Medical Research Council in Cambridge, England spent several years examining how proteins bind. They generated a huge number of mutant proteins with random amino acids confined to coherent patches. Despite having deliberately guided the mutations toward an intended goal, they found that a library containing between 10 million and 100 million variants is needed in order to get a particular protein to bind to another one with at least modest strength.29,30 Behe estimated that this is statistically equivalent to requiring a precise residue in a specific location five or six times (205 or 206).31
We saw above that mutations from all organisms which ever lived would provide fewer opportunities than necessary to generate a four specific amino acid pattern. Note that not only must stable binding occur, but it must also be restricted to the proteins which jointly provide a useful biological function. Less demanding interactions, using fewer amino acids would generally be too weak, and would lead to many interfering wrong interactions. In addition, there are many positions on a protein’s surface at which weak binding could occur, so the potential for incorrect associations would be immense.
Bruce Alberts, former president of the US National Academy of Sciences has pointed out that nearly every major process in a cell requires assemblies of 10 or more protein molecules.32 A mere binary interaction between proteins A and B would have required nature to generate and test about 1020 binding sites and a tertiary interaction A-B-C would have required testing about 1040 alternatives. Random mutations could not have produced anywhere near 10 correct protein-binding interactions.
The flagellum has dozens of protein parts that bind specifically to their correct partners; the cilium has hundreds. All the correct partners must associate, with binding to only suitable locations which generate the functional topology. In total there are thousands or tens of thousands of protein-protein binding sites in free-living cells.
Improbability as a post facto artefact?
One might object that an observed feature seems very unlikely to have arisen by chance, but unusual things do occur which had not been predicted in advance. Perhaps there are many random DNA patterns which offer opportunities for innovation and something useful was bound to happen. This is a poor argument. How often has inserting random DNA into a genome produced new useful functions? Let’s examine this objection more closely.
There are many examples of cellular features which permit little sequence variability. Examples include extreme protein invariability,33 or mandatory perfect 7–8 nt miRNA:mRNA pairing to hundreds or thousands of mRNA partners. Chance mutations will not produce a new miRNA seed region which just happens to be able to regulate the right ensemble of mRNAs, plus all the proteins needed.34 But the full picture is statistically worse. Dozens to thousands of different miRNA seed patterns each regulate a distinct collection of mRNAs, depending on the organism.35
As another example, one could argue that splicing out a spliceosomal intron might occur by chance, since evolution had not predicted this particular one had to be removed. Perhaps a multitude of other introns had gotten inserted at the same time. Consider, however, that a lineage having no or very few introns would have out-populated a seriously damaged lineage. Many or all introns would need to be removed at the same time at an evolutionary starting point to compensate for the burden of carrying all the additional spliceosome proteins.36 The correct consideration is not whether chance mutations could have removed a single damaging intron, but all of them.
Should one question evolutionary theory?
We should reject narratives in science, medicine, court cases, economics, and other areas if the theories are based on a series of statistically unrealistic ‘Insanely Improbable Chance Change’ (IICC) events. After this rather lengthy preparation we are now ready to review some of the tRNA features mentioned in parts 1–4.
Could a minimally viable tRNA subsystem have arisen naturally?
At some point in the evolutionary narrative there must have been an ancestral cell with the key features we observe today. This includes transcription, translation, an outer membrane, reliable energy production, metabolism, and reproduction. We’ll call this the ‘FUCA’ (First Universal Cellular Ancestor). This hypothetical entity would be as simple as possible but must be viable and fulfill the minimal requirements to be defined as a cell. The subsequent LUCA (Last Universal Common Ancestor) is not claimed to be minimally complex, and we are referring to a still earlier putative organism.37
If a FUCA never existed, then extant life could not have arisen from a common ancestor. We are not interested here in prebiotic or RNA world speculations preceding a FUCA. We are referring to an unavoidable stage in a conceptual evolutionary trajectory. It cannot be circumvented. This putative ancestor would have required dozens of functioning tRNAs to serve as translation adaptor molecules.
When might the tRNAs have arisen? There are only two possibilities. Either they arose exactly simultaneous with the FUCA, or before. By the definition of a FUCA, the tRNAs could not have evolved later. Any conceptual entity lacking a minimal tRNA-based translation would have been pre-FUCA.
Could tRNAs have arisen simultaneous with a FUCA?
Perhaps an ensemble of tRNAs appeared simultaneous with all the other parts necessary for a FUCA to be viable. This view would recognize the holistic nature of cells and how the subsystems are inextricably linked. The FUCA would reliably synthesize many copies of all necessary tRNAs (i.e. each with a suitable secondary structure and appropriate anti-codon). This scenario would not be consistent with naturalism, however. It would be indistinguishable from a miraculous event.
The chicken-and-egg dilemma would still be present, although now shortened timewise. The very first mRNA translated would have required functioning tRNAs. One cannot admit that tRNAs are a prerequisite for translation but claim that the FUCA manufactured them. This manufacturing process would require functioning transcription and biosynthesis, meaning the pre-existence of proteins which themselves depend on translation using tRNAs. Therefore, a naturalist should reject the first conceptual possibility, but not a creation scientist.
Could tRNAs have arisen before a FUCA?
If the first FUCA did not immediately begin manufacturing tRNAs, then these would have had to already exist and somehow been made available to a FUCA’s ancestor. Admittedly, this poses major difficulties, but this is the only alternative available. A constant supply of tRNAs would have been necessary for translation during the pre-FUCA period. This strategy would have to then be replaced in the FUCA by DNA which encodes these tRNAs plus all the features to transcribe them. The entire chain of evolutionary intermediates would have to occur with no intelligent guidance and all the stages would need to remain viable. Is this reasonable?
Long RNA single-strand sequences will form secondary structures whenever intramolecular folding brings regions of base-pairing counterparts into contact. In part 2, we showed that tRNAs will not fold into the necessary secondary structures without dozens of additional biochemical modifications. Some modifications prevent incorrect base pairings, and others are needed for correct pairings to be possible. This poses a chicken-and-egg dilemma, since the prerequisite enzyme proteins cannot be manufactured without functional tRNAs.
Postulating a naturalistic origin of the complex aminoacyl-tRNA synthetases and ribosome molecular machines is already unreasonable. But now we see in addition that the predecessor tRNAs would lack the necessary three-dimensional structural integrity for these to operate on and with. Extraordinary structural precision on parts of tRNAs are necessary to avoid mischarging activated tRNAs and to avoid translation reading frame errors.
A naturalist is now confronted with a dilemma. Either reject the first scenario based on philosophical preference or reject the second scenario based on sound logic.
Evolution of additional improvements by tRNAs
Some naturalists may wish to ignore the lack of a feasible starting point for tRNAs in a FUCA but argue that subsequent evolutionary innovations could occur naturally. Theistic evolutionists might claim this. Other worldviews postulate occasional divine miracles followed by unguided evolutionary processes. We will review some of the processes tRNAs participate in to judge the merit of these views.
Examples requiring more than four precise mutations on contiguous positions
The study referred to in figures 3 and 4 of part 4 documents how ~44 genes are regulated by a specific tRNA fragment, tRF5-GluCTC.38 A specific 17-nt perfect adjacent base-pairing patch (plus several more in another region) is necessary to target the mRNA of APOER2, a cell surface receptor involved in signal transduction and endocytosis. We proposed that the RSV virus might have been designed to stimulate production of tRF5-GluCTC and thereby regulate these 44 genes. The more than 17 perfect pairs between tRF5-GluCTC and the mRNA of the APOER2 protein could not realistically be attributed to random mutations. Claiming a correct group of 44 genes then became properly regulated through chance mutations pushes credibility into absurdity.
In another study discussed in part 4, several Drosophila genomes possess some 7-nt regulatory target sites. These are perfectly complementary to the respective 5’tsRNA (tRNA-derived small RNAs), so mutations at precise locations would have been necessary.39,40 In addition, special mutations would have been needed to produce the proteins involved in the processing steps. All of this would have had to occur during a short evolutionary time slot using far less than 1040 mutating organisms.
We mentioned in part 4 that many cases are known of tsRNAs functioning like miRNAs, having perfect seed regions of about 7 nucleotides (plus additional nt patterns necessary for accurate target recognition).
The tRF (tRNA-derived fragment) interactions with key proteins of the translation machinery also rely on sequence-specific binding, whether ribosomal proteins or translation initiation factors.39
The limit to four precise mutations or their statistical equivalent was based on an upper limit of how many organisms could have evolved over 10 billion years. The above examples are found in multicellular organisms with considerably lower Ne than bacteria and a small fraction of the putative available evolutionary time. There is, however, a small mathematical detail we should not overlook. Some of the necessary nucleotides will match the needed position by chance.
There is a 1 in 4 chance per position to produce a base-pairing interaction. For seven positions, about 7 × 0.25 = 1.75 nucleotides will be correct by chance. Countering this objection is the high probability that binding to a random mRNA or other partner would be deleterious, downregulating the wrong molecule and tying up a reactant needed for the correct function. Therefore, many more precisely located mutations would be necessary to find a beneficial evolutionary starting point which targets only, or primarily, the correct partners.
Examples requiring more than the equivalent of four precise mutations
In parts 1–4 we discussed many chemical modifications and quality control processes. These rely on binding patterns and novel proteins which jointly require more than the equivalent of four precise mutations. Often a unique enzyme is necessary. Protein families are extremely rare subsets in protein sequence space,41 and obtaining one or more new proteins would have required far more than the statistical equivalent of four precise mutations. We can state this confidently knowing the difficulty of also obtaining stable protein-protein bindings, and to only correct partners. tRNA-type specific ribonucleases (special proteins) are so discriminating they can identify malformed tRNAs and degrade them.
In part 3, quality control mechanisms designed to identify and degrade flawed tRNAs were described. Special protein-based molecular machines are necessary and feedback loops exist. For example, build-up of uncharged tRNAs communicates that protein translation should be slowed down. This is actively executed by inhibiting MEK2 kinase activity and by regulating the translation initiation factor eIF2α.1
In part 4, we discussed many processes tRNAs and their derivates are involved in. Under stress or nutrient limitation, the concentration of uncharged tRNA increases. This initiates a biochemical process that slows down translation.42 We also saw that many tRNAs are involved in other pathways which are not related to genetic code translation,43 and for strand transfer during retroviral replication.43
tRNAs are also involved in the first step of heme and chlorophyll biosynthesis, are employed as reverse transcription primers, participate in targeting proteins for degradation via the N-end rule pathway44 (which helps regulate protein concentrations45), and participate in regulated cell death (apoptosis).1
We also documented that tRNA degradation fragments are not random RNA but carefully crafted by special enzymes which are often tRNA-type specific. These special fragments are involved in cell-to-cell communication, immune signalling, and cell-state transitions. They play a role in suppressing tumour formation and metastasis and can regulate translation by displacing initiation factors. They are involved in modifying chromatin organization, regulating apoptosis and cell growth, maintaining epigenetic inheritance in germ-line cells, suppressing movement of transposable elements, and they interact with the immune system.
We also mentioned in part 4 that higher levels of uncharged tRNAs prevent tRNAMet from being processed by the ribosome. This is beneficial for the cell and occurs thanks to a reliable interaction with GCN2 kinase. From an information processing standpoint, a ‘variable’ (a specific site on GCN2) is assigned a Boolean value (e.g. ‘tRNA is attached’). (‘Variables’ are implemented by cellular codes using specific binding locations and patterns.46) The modified GCN2 then interacts with a specific location on the ribosome eIF2α factor (which is another variable) to phosphorylate it. This inactivates it, thereby preventing the initiator tRNAMet from forming.
At some point in the evolutionary model, various classes of tsRNA fragments would have had to be generated by novel enzymes. These would function in new processes, such as in regulating the concentration of mRNAs and miRNAs, in processes to hinder cancerous growth, and in suppressing metastasis. tiRNAs (tRNA-derived stress-induced RNAs), would also would have had to develop the ability to act as signalling molecules that participate in cell-to-cell communication, in immune signalling, and as biomarkers to detect stress-induced tissue damage.
Also mentioned in part 4, tsRNAs can regulate translation in various ways, for example by displacing translation initiation factors, or by displacing mRNA from the translation initiation complex. A specific fragment, the 18 nt 5′tRF can only affect translation if a specific uridine is first enzymatically transformed to pseudouridine (Ψ). Some tRFs can autoregulate by binding to the aminoacyl-tRNA synthetases involved in producing its parent tRNA.47,48
tRFs are also known to be involved in: modifying chromatin organization; regulating apoptosis and cell growth; epigenetic inheritance affecting offspring; suppressing movement of transposable elements; and interaction with the immune system.
Integration of subsystems with multiple cellular processes is often observed in the cellular design. For example, aminoacylated tRNAs (aa-tRNAs) are also involved in non-ribosomal peptide bond formation, post-translational protein labelling, modification of phospholipids in the cell membrane, and antibiotic biosynthesis.49
We can only mention here two other mathematical approaches researchers have used to show the inadequacy of evolutionary theory. One approach is to consider the entire ‘reservoir’ of mutations which could have occurred and demonstrate this is insufficient to explain all biological features which exist.50,51 We have shown that 1040 mutational events could not possibly explain the ‘simplest’ subsystem of the genetic code.
The second approach considers what Sanford and colleagues call the ‘waiting time problem’.52 The features we mentioned in the tRNA subsystem which require more than 4 precise mutations, or the statistical equivalent, could not have arisen in 10 billion years of evolution, far less all biological features which have existed.
We conclude that evolution is inadequate to explain the existence of a minimally functional ensemble of tRNAs needed by the genetic system, and also inadequate to explain any putative fine-tuning of the processes tRNAs and their fragments are involved in should such an initial state have arisen.
References and notes
- Truman, R., The surprisingly complex tRNA subsystem: part 1—generation and maturation, J. Creation 34(3):80–86, 2020. Return to text.
- Wahl, M.C., Will, C.L., and Lührmann, R., The spliceosome: design principles of a dynamic RNP machine, Cell 136:701–718, 2009. Return to text.
- Shi, Y., Mechanistic insights into precursor messenger RNA splicing by the spliceosome, Nat. Rev. Mol. Cell Biol. 18:655–670, 2017. Return to text.
- Wilkinson, M.E., Charenton, C., and Nagai, K., RNA splicing by the spliceosome, Annu. Rev. Biochem. 89:1.1–1.30, 2020. Return to text.
- Ramakrishnan, V., Ribosome structure review and the mechanism of translation, Cell 108:557–572, 2002. Return to text.
- Rajendran, V., Kalita, P., Shukla, H., Kumar, A., and Tripathi, T., AminoacyltRNA synthetases: structure, function, and drug discovery, International J. Biological Macromolecules 111:400–414, 2018. Return to text.
- Ibba, M. and Söll, D., The renaissance of aminoacyl-tRNA synthesis, EMBO Rep. 2(5):382–387, 2001. Return to text.
- Ibba, M. and Söll, D., Aminoacyl-tRNA Synthesis, Annual Review of Biochemistry 69:617–650, 2000. Return to text.
- Rubio Gomez, M.A. and Ibba, M., Aminoacyl-tRNA synthetases, RNA 26:910–936, 2020. Return to text.
- Truman, R., The surprisingly complex tRNA subsystem: part 2—biochemical modifications, J. Creation 34(3):87–94, 2020. Return to text.
- Truman, R., The surprisingly complex tRNA subsystem: part 3—quality control mechanisms, J. Creation 35(1):98–103, 2020. Return to text.
- Truman, R., The surprisingly complex tRNA subsystem: part 4—tRNA fragments regulate processes, J. Creation 35(1):104–110, 2020. Return to text.
- The probability of transition mutations (A ↔ G or C ↔ T) differ somewhat from transversion mutations (A or G are converted ↔ T or C), but this does not affect the essence of our discussion. Sometimes necessary mutations could be more likely, other times less likely. Return to text.
- Crooks, G.E., Hon, G., Chandonia, J.M., and Brenner, S.E., WebLogo: A sequence logo generator, Genome Research 14:1188–1190, 2004. Return to text.
- Qu, W., Cingolani, P., Zeeberg, B.R., and Ruden, D.M., A bioinformatics-based alternative mRNA splicing code that may explain some disease mutations is conserved in animals, Front. Genet. 8(38):1–12, 2017. Return to text.
- Lukasik, A., Wójcikowski, M., and Zielenkiewicz, P., Tools4miRs—one place to gather all the tools for miRNA analysis, Bioinformatics 32(17):2722–2724, 2016. Return to text.
- Wang, R., Wang, Z., Wang, J., and Li, S., SpliceFinder: ab initio prediction of splice sites using convolutional neural network, BMC Bioinformatics 20(652):1–13, 2019. Return to text.
- Behe, M.J., The Edge of Evolution: The search for the limits of Darwinism, Free Press, New York, 2008. Return to text.
- White, N.J., Antimalarial drug resistance, J. Clin. Invest. 113:1084–92, 2004. Return to text.
- Bray, P.G., Martin, R.E., Tilley, L., Ward, S.A., Kirk, K., and Fidock, D.A., Defining the role of PfCRT in Plasmodium falciparum chloroquine resistance, Mol. Microbiol. 56:323–333, 2005. Return to text.
- Kublin, J.G., Cortese, J.F., Njunju, E.M., Mukadam, R.A., Wirima, J.J., Kazembe, P.N., Djimde, A.A., Kouriba, B., Taylor, T.E., and Plowe, C.V., Reemergence of chloroquine-sensitive Plasmodium falciparum malaria after cessation of chloroquine use in Malawi, J. Infect. Dis. 187:1870–1875, 2003. Return to text.
- Whitman, W.B., Coleman, D.C., and Wiebe, W.J., Prokaryotes: the unseen majority, PNAS 95:6578–6583, 1998. Return to text.
- Behe ref. 18, chapter 5. Return to text.
- Eyre-Walker, A. and Keightley, P.D., The distribution of fitness effects of new mutations, Nature Reviews Genetics 8:610–618, 2007. Return to text.
- Robert, L., Ollion, J., Robert, J., Song, X., Matic,I., and Elez, M., Mutation rates and effects in single cells, Science 359(6381):1283–1286, 2018. Return to text.
- Patwa, Z. and Wahl, L.M., The fixation probability of beneficial mutations J. R. Soc. Interface 5(28):1279–1289, 2008. Return to text.
- Bobay, L.-M. and Ochman, H., Factors driving effective population size and pan-genome evolution in bacteria, BMC Evol Biol. 18:153, 2018. Return to text.
- Carter, R., A successful decade for Mendel’s Accountant, J. Creation 33(2):51–56, 2019. Return to text.
- Griffiths, A.D. et al., Isolation of high affinity human antibodies directly from large synthetic repertoires, EMBO J. 13:3245–3260, 1994. Return to text.
- Smith, G.P. et al., Small binding proteins selected from a combinatorial repertoire of knottins displayed on Phage, J. Mol. Biol. 277:317–332, 1998. Return to text.
- Behe, ref. 18, p. 132. Return to text.
- Alberts, B., The cell as a collection of protein machines: preparing the next generation of molecular biologists, Cell 92:291–293, 1998. Return to text.
- For example, the H4 protein sequences are virtually identical for all organisms. See Piontkivska, H., Rooney, A.P., and Nei, M., Purifying selection and birth-and-death evolution in the histone H4 gene family, Molecular Biology and Evolution 19(5):689–697, 2002. Return to text.
- O’Brien, J., Hayder, H., Zayed, Y., and Peng, C., Overview of MicroRNA biogenesis, mechanisms of actions, and circulation, Frontiers in Endocrinology 9(402):1–12, 2018. Return to text.
- miRBase: the microRNA database, mirbase.org/. Return to text.
- Matera, A.G. and Wang, Z., A day in the life of the spliceosome, Nature Reviews Molecular Cell Biology 15(2):108–121, 2014. Return to text.
- Theobald, D.L., A formal test of the theory of universal common ancestry, Nature 465(7295):219–222, 2010. Return to text.
- Deng, J. et al., Respiratory syncytial virus utilizes a tRNA fragment to suppress antiviral responses through a novel targeting mechanism, Mol Ther. 23:1622–1629, 2015. Return to text.
- Luo, S., He, F., Luo, J., Dou, S., Wang, Y., Guo, A., and Lu, J., Drosophila tsRNAs preferentially suppress general translation machinery via antisense pairing and participate in cellular starvation response, Nucleic Acids Res. 46:5250–5268, 2018. Return to text.
- Jehn, J. and Rosenkranz, D., tRNA-derived small RNAs: the good, the bad and the ugly, Med One 4:e190015, 2019. Return to text.
- Axe, D.A., Estimating the prevalence of protein sequences adopting functional enzyme folds, J. Mol Biol. 341(5):1295–1315, 2004. Return to text.
- Pan, T., Modifications and functional genomics of human transfer RNA, Cell Res 28:395–404, 2018. Return to text.
- Schimmel, P., The emerging complexity of the tRNA world: mammalian tRNAs beyond protein synthesis, Nat. Rev. Mol. Cell. Biol. 19:45–58, 2018. Return to text.
- The amino acid residue used at the N-terminal end of a protein serves as a signal for the intended half-life, in bacteria and eukaryotes. Ubiquitin ligases and other enzymes then mediate the degradation. Return to text.
- Hopper, A.K., Transfer RNA post-transcriptional processing, turnover, and subcellular dynamics in the yeast Saccharomyces cerevisiae, Genetics 194:43–67, 2013. Return to text.
- Truman, R., Cells as information processors—part 1: formal software principles, CRSQ 52:275–309, 2016. Return to text.
- Kim, H.K., Transfer RNA-derived small non-coding RNA: dual regulator of protein synthesis, Mol. Cells 42(10):687–692, 2019. Return to text.
- Qin, C., Xu, P.-P., Zhang, X., Zhang, C., Liu, C.-B., Yang, D.-G., Gao, F., Yang, M.-L., Du, L.-J., and Li, J.-J., Pathological significance of tRNA-derived small RNAs in neurological disorders, Neural Regen Res 15(2):212–221, 2019. Return to text.
- Kanai, A., Welcome to the new tRNA world! Frontiers in Genetics 5(336):1–2, 2014. Return to text.
- Truman, R. and Borger, P., Genome truncation vs mutational opportunity: can new genes arise via gene duplication?—part 1, J. Creation 22(1):99–110, 2008. Return to text.
- Truman, R. and Borger, P., Genome truncation vs mutational opportunity: can new genes arise via gene duplication?—part 2, J. Creation 22(1):111–119, 2008. Return to text.
- Sanford, J., Brewer, W., Smith, F., and Baumgardner, J., The waiting time problem in a model hominin population, Theoretical Biology and Medical Modelling 12:18, 2015. Return to text.





Readers’ comments
Comments are automatically closed 14 days after publication.