
Responding to supposed refutations of genetic entropy from the ‘experts’

The idea of genetic entropy (GE) was pioneered by retired Cornell professor Dr John Sanford. The basic idea is that most mutations have such a small effect that they cannot be effectively removed by natural selection. Thus, as mutations build up in the genome, the net result should be genetic decay. The concept is simple enough. It was outlined in a popular book titled Genetic Entropy, but it is also supported by rigorous computer simulations using a powerful program called Mendel’s Accountant and real-world examples.
There are several ways in which evolutionists attempt to evade the effects of genetic entropy. Objections have included appeals to truncation selection, synergistic epistasis, mutation counts, and debates over the ‘true’ mutation spectrum. These have been analyzed separately and in combination and found to be wanting.
The evolutionary community has not given in. Several attempts have been made to discredit GE, but these efforts have not been strong. Basic misunderstandings of what GE claims, what evolution claims, and the power of the respective alternatives abound. For example, see Critic ignores reality of Genetic Entropy. The debate has continued on Dr. Joshua Swamidass’ Peaceful Science blog. He is an evolutionist and the author of The Genealogical Adam and Eve (GAE), which received a scathing review on Creation.com. We find his science to be not at all ‘peaceful’, but openly hostile to the views of biblical creationists. In fact, he openly admits that his GAE hypothesis represents a sort of Hegelian1 synthesis between creationism and evolutionism.2 Readers be cautioned! Tricky rhetorical flourishes are not good arguments, but they abound in those pages.
What follows is a summary of six arguments against GE recently made at Peaceful Science, followed by our answers. The references are to forum posts (sub-posts within longer discussions). Each statement will be documented in the footnotes in case these links eventually change or disappear. A disclaimer is also appropriate: we have done our best to give accurate summaries of the intended arguments of these experts, but often we have had to piece together responses from different places, as the intended meanings were often shrouded in opaque or indirect language. Also, we removed paragraph breaks to save space in the footnotes while preserving the original wording. In fairness to them, please note that the statements they made were not meant for publication in a scientific context. Thus, the grammar may be a little loose, which fits with the blog format.
We also checked with Dr Sanford to see if he had anything else to add after this article was written. In each of the following sections he appended a note. Here are his first comments:
The ‘experts’ mentioned below are very well-credentialed scientists. Yet, they are not experts on the specific topic at hand. They have not spent the last twenty years studying the problem of mutation accumulation. Their six objections below are just ‘off the cuff’ statements that do not seem to reflect careful consideration. They naturally feel like they need to say something, but they only offer vague and dismissive comments. If this is all they have, I am amazed, and very encouraged!
- Mutations & Equilibrium
Claim: As the load of deleterious mutations grows over time, the pool of possible beneficial mutations also grows with it. This eventually leads to an equilibrium, preventing fitness decline beyond a certain point.
This was stated by Dr. Daniel Stern Cardinale, evolutionary biologist at Rutgers University,3 and by Dr. Joseph Felsenstein,4 population geneticist at the University of Washington and author of the popular PHYLIP evolutionary tree-building software.5 Dr. Swamidass has also stated this objection.6
Answer: Mutation-drift equilibrium is a standard part of many evolutionary models. Given many millions of years, one would expect genomes to become saturated with mutations, reaching an equilibrium where the number of new mutations is balanced by the number of mutations lost through random genetic drift and purifying selection. However, this is theory only. Mendel’s Accountant has shown us that extinction should happen long before mutation-drift equilibrium is reached. The sheer number of mutational possibilities argues against the idea that back mutations can come in for the rescue. Given the number of variants so far observed in the human genome, and given the fact that the great majority of these are extremely rare, it is clear that there are many more possible mutations in the genome than are commonly found. Even if a mutation were to happen in the same location, the probability of switching back to the original letter is but 1 in 3.
Worse, not all mutations have the same probability. If a C→T mutation (one of the most common mutations we see) happens, the reverse [T→C] is less likely to occur. Thus, while we are waiting for our rare T→C back mutation to happen, other C→T mutations should be occurring in the near vicinity. The same is also true of A→G vs G→A, where the former is much more common than the latter. This is all based on simple chemistry. But these are all examples of transition mutations, where a swap has been made between a purine and a purine (A and G) or a pyrimidine and a pyrimidine (C and T). Transversion mutations (swapping a purine for a pyrimidine and vice versa) are even more rare, so while the organism is waiting for a transversion to reverse itself, transition mutations (mostly C→T and A→G) will be happening all around it.
We can use a simple word analogy here. Start with the word HOUSEHOLD and add a typo to make it BOUSEHOLD. An intelligent mind will immediately see the problem and correct the mistake. But what about unguided chemistry? First, the system would have to recognize there is a problem, and biology has no way to do that. If an organism dies because of a genetic spelling error, it is dead and cannot correct the problem. If an organism lives with a spelling error, it does not know that a problem exists. If the organism got really lucky and a back mutation happened that fixed the wording, all would be well. But what are the chances? It does not know there is a problem in general, and it cannot know which letter is the problem even if it knew there was a spelling error in that one word. Would another mutation ever be expected to fix it when there are so many other possibilities, like MOUSEHOLD? And what would happen if a few more mutations happened before the fortuitous back mutation arose? We might go from BOUTNHELDE to HOUTNHELDE, which is unhelpful. Back-mutations are only helpful if the original context is preserved.
Finally, in a mutation-saturation scenario, one would expect to see many places in the genome where all four letters are found (A, C, G, and T). The human genome is far from that position.
Comments from Dr Sanford:
This argument is: 1) merely dismissive, 2) categorically wrong, and 3) without a rational or data-driven basis. Obviously, rapidly accumulating deleterious mutations do not lead to more and more beneficial mutations. Rather, the much more abundant deleterious mutations effectively overwhelm and negate the fitness effects of the extremely rare beneficial mutations. The ratio of bad to good mutations is, minimally, 1000:1. With or without selection, bad mutations will always accumulate much more rapidly that beneficial mutations. We have done thousands of numerical simulations showing this. Even given the most generous parameter settings, the near-neutral bad mutations consistently accumulate about 1000 times faster than the beneficial mutations.
- Natural selection equilibrium
Claim: Natural selection will kick in after a certain amount of near-neutral mutations have accumulated. Effectively neutral mutations may well accumulate, but once they start to create enough damage to cause reproductive problems, they are no longer effectively neutral. At this point, they become selectable and NS will prevent further accumulation (again, leading to an equilibrium).
This was stated by Stern Cardinale,7 by Dr. Stephen Schaffner,8 population geneticist and computational biologist at the Broad Institute of MIT and Harvard,9 and (possibly less clearly) by Felsenstein.10
Answer: This is essentially the ‘mutation count’ hypothesis. It has already been answered.11 But Stern Cardinale also claimed, “Dr. Sanford’s model requires mutation accumulation without purifying selection…” The near-neutral mutations do add up and are a factor in reproductive success. But since a near-neutral on its own does little to influence reproduction, they end up acting as if they were completely neutral and thus are only affected by genetic drift. And yet, as we will see in point #3 below, these adversaries want to invoke as many positive near-neutral mutations as there are deleterious near-neutral mutations, which is entirely odd. How can random changes to an information system be good for that system as often as they are bad? That’s like saying spelling errors are as likely to improve this article as they are to detract from it. Also, appeals to natural selection preserving the “least bad” genotypes are missing the mark. The point of genetic entropy is that the “least bad” in one generation is actually a little “more bad” than in the generation before. As slight mutations build up, the fitness of all organisms in the population should decline.
Comments from Dr Sanford:
They have not carefully thought through what they are saying. The nature of near-neutral mutations is such that they are not only un-selectable due to environmental noise, but they are also un-selectable because they are ‘noise’ to each other. Thus, as the number of neutral mutations accumulate, selection gets worse, not better. If an individual carries just one near-neutral mutation, it might be very weakly selectable (but probably not, as environmental noise will override its tiny effect, so there will be little or no selection at all). If each individual has 10,000 near-neutrals, selection has to try and select for (or select against) all 10,000 conflicting mutational fitness effects simultaneously. Ten thousand independent mutational fitness effects (usually bad ones, vanishingly few good) will not just be pulling in different directions with each other, they will all act as ‘noise’, blotting out the fitness effects of each other. Haldane makes it clear that only a few mutations can be effectively selected for simultaneously. Trying to select for too many mutations at once totally overwhelms any type of selection. Indeed, selection interference not only prevents selection for countless near neutrals, it even interferes with selection for the more impactful mutations that are also accumulating.
- The distribution of fitness effects for nearly neutral mutations is balanced
Claim: All our data on mutations apply only to mutations of large enough effect to be measured and studied, and these mutations are of the selectable, not the nearly neutral, variety. Therefore, we know nothing about the distribution of fitness effects for nearly neutral mutations. They are as likely to be beneficial as they are to be deleterious.
This was stated by Schaffner.12 Both he13 and Felsenstein14 stated they are equally likely to be deleterious or beneficial.
Answer: Dr Sanford’s conceptual mutation spectrum was developed from the theories of neutral evolutionists like Kimura and Ohta. It was Kimura who said most mutations were nearly neutral, suggesting a distribution like this:15

Sanford added beneficial mutations to this. Note the curves on the left and right side of “0” have the same shape:
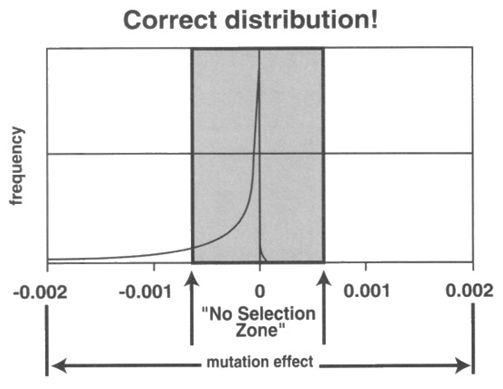
See Critic ignores reality of Genetic Entropy for more information.
Here is the distribution of effects Dr. Schaffner is arguing for:16

Dr Schaffner confirmed he agreed with what is drawn here. He acknowledges that selectable, ‘truly’ beneficial mutations are much rarer than selectable, ‘truly’ deleterious mutations. He maintains, however, that ‘effectively neutral’ mutations are equally likely to be beneficial or deleterious. He refused to answer when asked to explain why the same factors that caused selectable mutations to be skewed toward deleterious would not also apply to neutrals as well. Worse, he is essentially confirming Sanford’s model, where deleterious mutations outweigh beneficial ones and where most mutations have little effect. Yet, this model could be studied with Mendel’s Accountant (hint: we pretty much already know what will happen).
The selection threshold, that is, the strength a mutation must have before it can be ‘seen’ by natural selection, has been quantified, for the first time, using Mendel’s Accountant.17 Any claim that new mutations are equally as likely to be beneficial as they are deleterious flies in the face of both expectation and measurement. The human genome demonstrates a huge load of rare, recessive, and deleterious mutations. These have been preserved by our rapid population growth. In rapidly expanding populations, new mutations are less likely to be lost, so they show us something more closely approximating the real mutation spectrum. We have also been able to see the effects of mutation accumulation on the human H1N1 influenza virus. After nearly 100 years in circulation, it became less deadly and less infectious as the number of spontaneous, random mutations climbed inexorably. At the same time, codon usage randomized, and nucleotide composition began to approach thermodynamic equilibrium. In fact, long-term mutation accumulation patterns followed a predictable pattern that closely matched the chemical probabilities of the various mutations. That shows us selection was not a significant factor in affecting which mutations accumulated.
How can they say we don’t know the distribution of fitness effects when we can certainly know what mutations are accumulating in genomes over time? Evolution cannot handle this. By definition, natural selection must be able to overcome the effects of thermodynamic pressures in chemistry. If not, all life would have already devolved into simple chemicals. Sanford and colleagues also studied the effects of strongly beneficial mutations. The models indicated that selection for a mutation also carries along the nearby deleterious alleles. Thus, ‘linkage’ is a major problem. In order for their idea to work, beneficial mutations must vastly outnumber deleterious ones.
Comments from Dr Sanford:
Again, they have not thought this through very carefully. If a mutation has any effect at all, it is because it is affecting some aspect of the organism’s biological information system. This is logically true whether a mutation is high impact, moderate impact, or nearly neutral. Random mutations in the genome are just like random letter changes in an instruction manual, or the random flipping of binary bits in computer code. In all these examples, we know for sure that random changes will always lead to a net loss of information, and almost all changes will be deleterious. Waiting for a beneficial mutation, even a near-neutral beneficial mutation, is like waiting to win a lottery. It should be obvious to any thinking person that near-neutral mutations, like all random changes in code (large or small), will very consistently be harmful.
- Mutation accumulation is not a problem for most species
Claim: Mutation accumulation may be a problem for humans, but there is no evidence that it represents a problem for any other life forms.
This claim was made by Felsenstein.18
Answer: The theory of genetic entropy was developed for complex, long-lived organisms that have long generation times and small populations. The only possible species that might escape it are bacteria. Thus, things like humans and elephants are the primary focus. However, this does not mean that smaller animals like mice or beetles escape. It might take longer for some species more than others to decline because of GE, but extinction is guaranteed if the mutation accumulation rate, population wide, is positive over time. It is true that humans carry a large load of deleterious mutations, and it is true that a large part of this is the huge increase in our population size in the recent past. Yet, if we were a much smaller population, we would still be accumulating mutations, and these would still have to be removed. Mendel’s Accountant has shown us how difficult it is to do this effectively, given the limited reproductive output of humans. There are both theoretical and computational arguments against long-term evolutionary population survival for humans, and there is good reason to think this would apply to other long-lived organisms with complex genomes. Thus, even though the mutational load on humans has increased drastically over the last few thousand years due to population growth, this does not mean that these ideas do not apply to other species. The fact that species have managed to survive this long is a testimony to the genius of the Creator, who designed living things with an amazing robustness and resistance to injury.
Comments from Dr Sanford:
Again, they have not thought this through very carefully. The mutation accumulation problem is most severe in small populations (because selection becomes less effective). There are almost eight billion people alive today, making us one of the largest mammalian populations. Any mammalian population with fewer individuals should be even more subject to genetic entropy than mankind. Clearly, population size is a significant factor in many past extinctions (due to mutation accumulation), and mutational meltdown is certainly relevant to all currently endangered species. Most mammals have mutation rates similar to humans. It is true that we take care of the old and sick, which may have a limited effect on selection efficiency. Then again, we tend to kill off a lot of people as we fight with each other. It should be obvious that genetic entropy is not limited to mankind.
- Junk DNA
Claim: Genetic entropy only works by presupposing a perfect genome but much of it is really random junk. Most mutations are deleterious only for organisms that are already well-adapted. If you presuppose that God created a perfect genome, then naturally any changes will be bad. But real life doesn’t work that way, and genomes were never perfect (or even largely optimal). Changes to the genome are as likely to be good as they are bad.
This was stated by Schaffner19 and Felsenstein.20
Answer: First, there is nothing in GE that requires the starting point to be ‘perfect’. You can begin with any arbitrary starting point and look at the direction of change from there (naturally, the general downward trend we observe does have implications about the starting state of the genome!). Mendel’s Accountant can be run for a specified time to allow for any amount of mutational ‘burn in’ the user desires. And, a set of pre-specified mutations, with any chosen distribution of mutation effects, can be front-loaded into any model run. Yet even when starting with a ‘perfect’ genome with no mutations at all, we consistently see degradation over time. Allowing burn-in or frontloading mutations only hastens extinction in model organisms.
Second, in Mendel’s Accountant, one can assume as much ‘junk DNA’ as one likes. This is easily accomplished by setting the mutation rate to a fraction of the expected real rate or by using a smaller model genome. Given an average of perhaps 100 new mutations per child per generation, what if only one of those was functionally important? Mendel’s Accountant can model that.
Third, the principle that ‘it’s always easier to break a machine than it is to improve upon it’ is by no means limited to perfect machines! This general principle applies to all functional machines—even the imperfect creations of humans. If you make unplanned and unguided changes to the blueprints of a 1960s Ford automobile (to use Schaffner’s example), you’ll find you’re much more likely to damage it than you are to improve upon it. In fact, the dramatic improvement in automobile reliability since the 1960s did not come from incremental changes but from dramatic, intelligently designed gestalts21 that came from human minds, like anti-lock brakes, on-board computers, and careful studies of metal corrosion when two dissimilar metals come into contact.
Comments from Dr Sanford:
They have not thought this through at all: 1) Genetic entropy does not presume that the genome started out perfect (although the Bible does indicate this). Rather, genetic entropy applies to any genetic system, young or old, perfect or flawed. Regardless of previous damage, deleterious mutations systematically take the whole system downward, not upward. 2) The ‘Junk DNA’ argument is an old rescue device; it is outdated, and it is increasingly being refuted. Why do they continue to cling to Junk DNA? The only reason hardcore Darwinists still cling so tightly to the idea that almost all the genome is junk is because, without it, they would have to admit genetic entropy is real. It has been conclusively established that a very large part of the human genome is functional. The enormous ENCODE project, plus multiple follow-on projects, make it clear that most of the genome has function. Researchers continue to discover more and more new functions within the DNA previously cataloged as ‘junk’.
- Allegations regarding research into the H1N1 virus by Sanford and Carter
Claim: Carter and Sanford are guilty of a fundamental error in methodology on their H1N1 research, rendering it inadmissible as evidence of genetic entropy.
John Mercer, a molecular biologist, referred to a figure from a paper co-authored by Carter and Sanford22 and stated, “In Fig. 2, H1N1 sequence differences are falsely described as ‘Relative mutation count.’ … The discontinuities are from rearrangements of segments, which are not mutations … Calling them all mutations is grossly false and incredibly misleading.”23
Answer: Being that one of the authors (RC) personally and painstakingly aligned those sequences, he should know what types of changes were occurring. Yes, there are discontinuities, which is why we separated the analysis into two main threads: the 2009-2010 pandemic viruses and the 1918-2010 H1N1 viruses that had infected humans and were not derived from the swine version. There are no major discontinuities among the viruses in the 1918-2010 set. If there were, they would have been easy to spot and the alignment would have become much more difficult. The influenza genome is composed of eight separate RNA segments. It is possible for RNA segments from one strain to ‘reassort‘ with RNA segments of another strain. If an animal is infected with two strains simultaneously, reassortment can occur as the RNA segments are packaged together in the viral envelope. This is apparently what led to the rise of the 2009 “swine flu” strain. Epidemiologists were afraid that this would reinvigorate the old H1N1 virus, and when it jumped to humans, the world got seriously worried. But these changes are obvious. Also, he used the word ‘rearrangement’ instead of the more correct ‘reassortment‘. It is unclear what he means. ‘Recombination‘ would exchange blocks of letters within the same RNA segment. Reassortment leads to the rise of new viral classes (e.g. H1N1, H3N2, H5N7, etc.). This is the basis of the rise of the major annual flu strains. But even without reassortment or recombination, deletions and smaller rearrangements can occur within the flu genome. Yet, there is extremely little non-coding RNA in that virus. There is little room for gross changes. Indeed, among the nearly 9,000 human and over 1,700 swine H1N1 sequences examined, nearly all mutations amounted to single-letter changes, small insertions, and small deletions.
Here is a screen shot of one of the worst sections in the alignment. This is part of the hemagglutinin (HA) gene. Strains from 1918 through 1936 are shown. The human and swine H1N1 reference genomes are also there. We see one three-letter deletion (keeping the downstream codons in-frame) and many single-nucleotide changes. There is no evidence for large-scale rearrangements, either within or among the eight segments of the H1N1 genome.

Being that we can see incremental changes from one virus to another when we order them in time, and being that there is a clear linear trend toward higher mutation counts over time, we were absolutely correct to use the phrase ‘relative mutation count’. Carter even provided an updated version of that image in More evidence for the reality of genetic entropy—update. In this image, viruses that infected humans are in black and those that infected swine are in grey. In later years, the swine version split into two major strains, as can be seen from the two parallel bands on the right side. However, the line representing the human version stayed tight and straight. The first break in the line was due to the human H1N1 disappearing in 1957. It was reintroduced in 1976 and circulated until 2009, when it went extinct again. The third section of black dots only shows us that the H1N1 virus circulating in humans today does not derive from the original human strain but from the 2009–2010 “swine flu” outbreak virus.
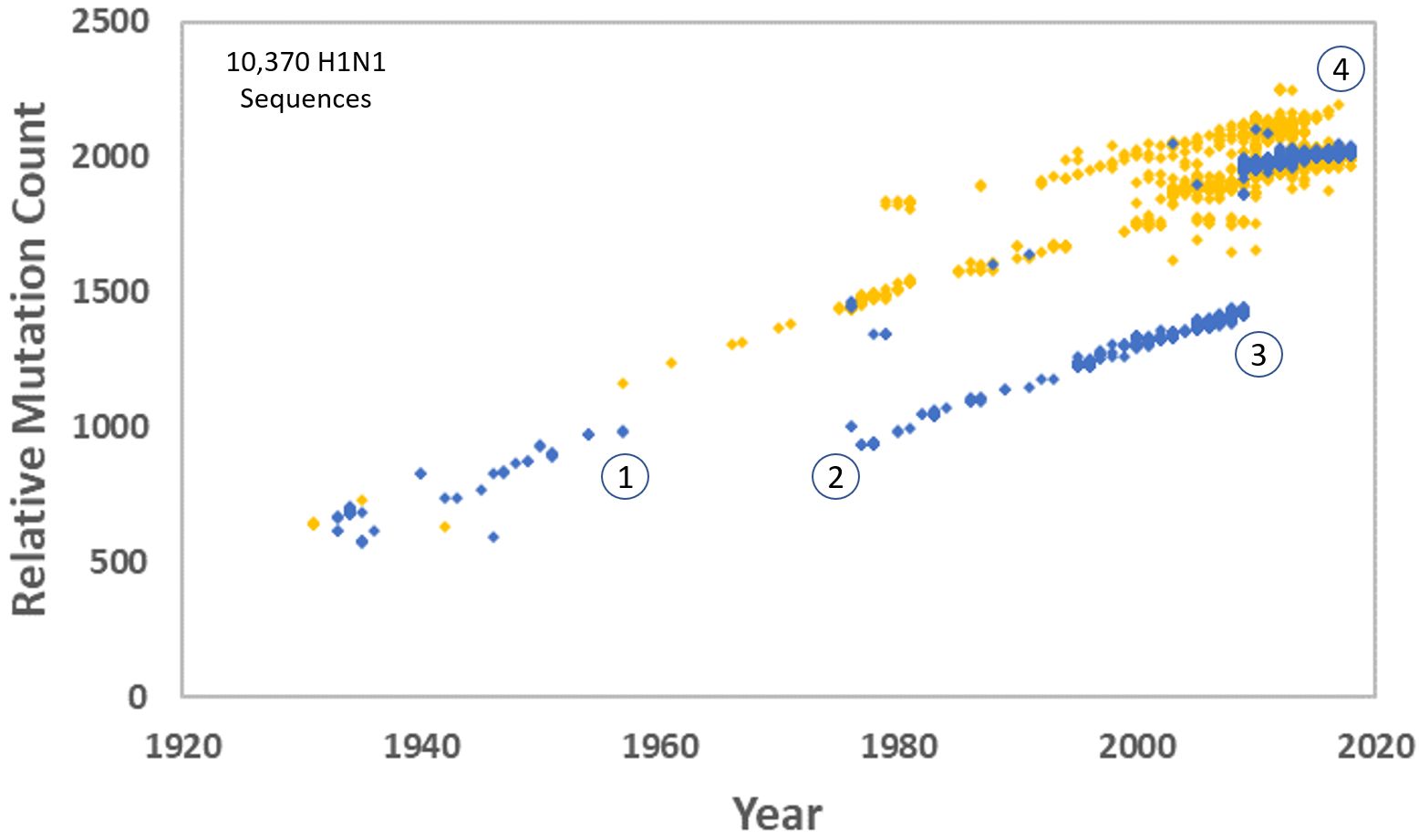
Using a pseudonym on Reddit (outside the Peaceful Science forum) Stern Cardinale added, “The 2009 pandemic H1N1 was reassorted from an ancestral human H3N2, not the previously circulating human H1N1. Which makes this paper even worse, since Carter and Sanford used the 2009 pandemic strain as their reference genome for the purposes of mutation accumulation. Which means even the most basic part of the study - how many mutations and which ones - is wrong.”24
Stern Cardinale fundamentally misunderstands what is going on. In our 2012 paper we specifically wrote, “The influenza genome consists of eight RNA segments totaling over 13,100 nucleotides. These code for up to eleven distinct proteins, two with alternate reading frames and one through alternate splicing. Each of the eight RNA segments has its own history of reassortment, inheritance, and mutation.” Clearly, we were well aware of the reassortment history among these viruses.
Thus, his claim that “The 2009 pandemic H1N1 was reassorted from an ancestral human H3N2, not the previously circulating human H1N1” is irrelevant. This was known to all parties.25
His following statement indicates more misunderstanding: “Which makes this paper even worse, since Carter and Sanford used the 2009 pandemic strain as their reference genome for the purposes of mutation accumulation.” Did we use the 2009 pandemic strain as a reference genome? Yes, when studying the pandemic strain viruses only. It would have been a mistake to use the 1918 virus to assess mutation accumulation rates if we were lumping the human version in with the pandemic swine virus. Why? Because they follow two distinct trendlines (see below). Instead, we compared apples to apples. Our analysis was entirely appropriate.

From the slope of the trend line, we concluded that the 2009–2010 pandemic H1N1 virus was picking up about 42 mutations per year but made no further comment. We then turned our attention to the 1918 H1N1 and its derivatives. After plotting all available H1N1 sequences, we noticed two different trend lines.
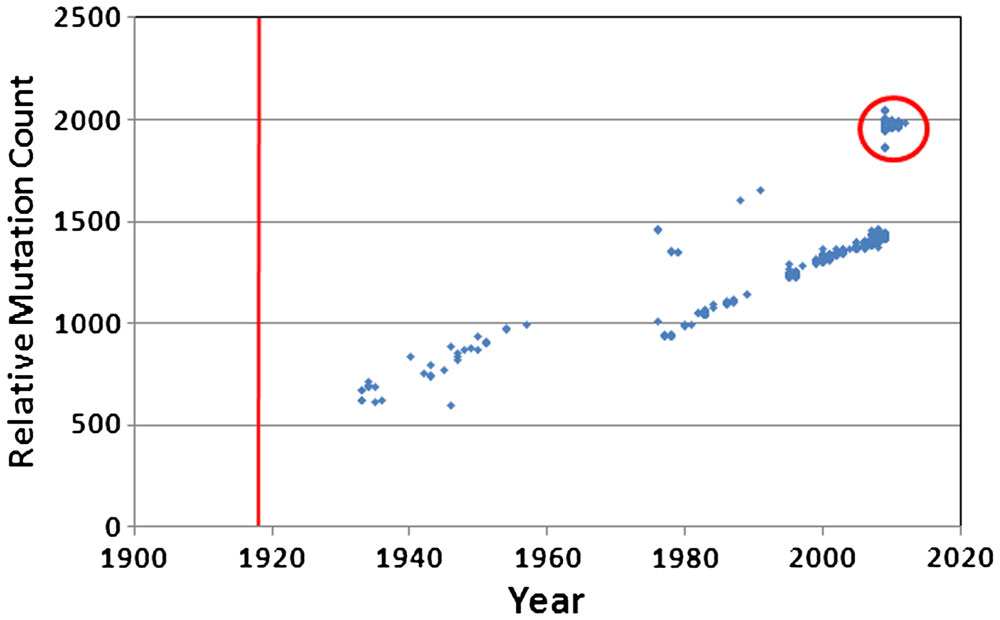
After removing the swine viruses, including the 2009–2010 pandemic viruses, and then adjusting for the 21-year gap, we were able to piece together the history of the original 1918 virus.

From the data we presented, we were able to discern that the virus had been picking up about 14 mutations per year of circulation, amounting to over 1,400 nucleotide changes (nearly 11% of the genome). We were also able to show that the genome was moving toward a state of maximum randomness. It was not becoming ‘better’. Instead, the laws of thermodynamics were driving it toward a state of maximum entropy, which is disallowed in the Darwinian model. Perhaps this is why they are trying so hard to discredit our work. The claim that the viral genome is trending toward randomness was documented in the original paper and reinforced with additional publications later.26,27
His final comment, “Which means even the most basic part of the study - how many mutations and which ones - is wrong,” is therefore shown to be completely incorrect.
Comments from Dr Sanford:
Carter can best respond to this, and he did above. I would just like to say that our original paper on the H1N1 virus is remarkably strong. There have been over 17,000 accesses of this peer-reviewed paper, and there has been no serious challenge to our work. There have only been a few pot-shots taken at our work from poorly informed and partisan people, such as seen here. Just cheap shots. Furthermore, our paper has direct relevance to COVID-19. Why do all pandemics peter out naturally and quite quickly? A strong case can be made for natural attenuation (an example of genetic entropy). Several current COVID-19 treatments now employ pharmaceuticals that accelerate RNA mutation rates, which is essentially accelerated genetic entropy.
Concluding Remarks
In reviewing the many attempted rebuttals from these various evolutionist experts, a few general observations can be noted. First, it often takes a lot of ‘doing’ to get any straight or direct answers as to why they reject Genetic Entropy. Second, we rarely see any evidence that these detractors have actually read Dr. Sanford’s book (as many of their objections are dealt with in the book itself) or any of the papers that have come via Mendel’s Accountant. Third, it is clear they oppose any challenge to Darwinism in principle. They take it as their ‘Primary Axiom’ and consider it unassailable. Finally, it is impossible to miss the fact that, even among the experts, there is no consensus as to why Genetic Entropy is supposed to be wrong. If you ask ten experts why they reject it, you’ll likely get ten different answers, often that contradict one another. This really is a huge ‘Achilles’ heel’ for evolutionary theory! Real science disproves Darwinian speculations. Attempts to show God’s design is not needed to explain the diversity of life on our planet all fall short.
References and notes
- The ‘Hegelian dialectic’ is a system of thinking that describes a thesis, which gives rise to a reaction (an antithesis), which leads to tension, that is eventually resolved by a synthesis of the two positions. This approach is the basis of Marxist philosophy, although it is certainly not restricted to these writers, having roots in Kant, Goethe, Spinoza, and others. Return to text.
- “In this case, [a Hegelian synthesis] is what the GAE will end up being :wink: . There are always hold outs for the thesis or antithesis, and that’s where the debates are loudest, but most people are not so invested to turn their back on a good synthesis.” discourse.peacefulscience.org/t/introducing-chad-from-middle-ground/11792/78. Return to text.
- “I have 100 lights that can be either red or green, and they randomly change back and forth at a constant rate. Right now, 90 are green, and 10 are red. From that specific state, how frequently will a green light turn red? 90% of the time. But what if 10 light turn red. Now there are 80 green lights and 20 red lights. In this new state, how frequently will a green light turn red? Now it’s about 80% of the time. What if it’s 50/50? Now they switch back and forth at equal frequency… My point is that if every mutation is either “green” or “red”, then as “red” mutations occur, the frequency of potential “red” mutations declines. So “red” mutations do not accumulate in a linear fashion. The answer to this problem is that there isn’t an answer; Sanford is just wrong.” discourse.peacefulscience.org/t/sft-on-genetic-entropy/11638/38. Return to text.
- “On theoretical grounds, we would expect that as the genome accumulates weakly deleterious mutations, the opportunity for them to be reversed by advantageous mutations would go up (just as the accumulation of minor and not disastrous typos in a book will increase the opportunity for advantageous typos).” discourse.peacefulscience.org/t/paul-price-what-are-the-substantive-critiques-of-genetic-entropy/11736/23, in his point #6. Return to text.
- en.wikipedia.org/wiki/Joseph_Felsenstein Return to text.
- “I think it is worth noting that the function is almost certainly not stable and fixed. Likely, as negative mutations accumulate, positive ones become more likely. As positive mutations accumulate, negatives ones become more likely. This might function somewhat like a thermostat. It would not require any special mechanisms, but be [sic] a consequence of the underlying fitness function and dynamics.” discourse.peacefulscience.org/t/the-distribution-of-the-effects-of-mutations/11869/30. Return to text.
- “Dr. Sanford’s model requires mutation accumulation without purifying selection , and also, ultimately, a decline in reproductive output due to that accumulation . This is simply contradiction; if the mutations in question result in a decline in relative reproductive success, then they are necessarily subject to purifying selection. I’ve heard and read the retort that the entire population is affected, so absolute reproductive ability declines, but relative output does not, so there can be no purifying selection. For this to be the case, there would have to be no differences in reproductive output across members of the entire population in question, despite each individual experiencing a unique set of mutations. Which, of course, is not a serious proposition. This means that selection can preserve the ‘least bad’ genotypes, if you will. Not pristine, not non-mutated, not optimal, but least bad.” discourse.peacefulscience.org/t/paul-price-what-are-the-substantive-critiques-of-genetic-entropy/11736/7. Return to text.
- “As you yourself have pointed out, there are far, far more sequence configurations that are suboptimal than there are optimal ones. For configurations that differ only by the kind of tiny fitness differences we’re talking about, the chance of the optimal configuration occurring by chance is exceedingly small. There is also (by definition) no way for natural selection to have generated the optimal configuration, since these differences are invisible to selection. Therefore, there is no reason to think the genome was ever in that optimal state. Instead, an actual genome is one randomly chosen configuration from among all of the selectively equivalent configurations, and effectively neutral mutations take move it to other selectively equivalent configurations. The precise fitness of the genome will drift up and down very slightly, but changes are as likely to be positive as negative. Note that this assumes a constant size for the population in question. If the population has gotten smaller, a new class of mutations becomes invisible to selection and these mildly deleterious mutations will accumulate until a new equilibrium is reached.” discourse.peacefulscience.org/t/sft-on-genetic-entropy/11638/247. Return to text.
- https://www.broadinstitute.org/bios/stephen-schaffner. Return to text.
- “Having far more deleterious than advantageous mutations does not predict that the same is true of substitutions in evolution. Population genetics calculations show that natural selection can discriminate against deleterious mutations, and do so very strongly.” discourse.peacefulscience.org/t/paul-price-what-are-the-substantive-critiques-of-genetic-entropy/11736/23, in his point #4. Return to text.
- Brewer, W., Baumgardner, J., and Sanford, J., Using numerical simulation to test the ‘Mutation-Count’ hypothesis; in: Marks II, R.J., Behe, M.J., Dembski, W.A., Gordon, B., and Sanford, J.C. (Eds.), Biological Information—New Perspectives, World Scientific, Singapore, pp. 298–311, 2013; worldscientific.com/doi/pdf/10.1142/9789814508728_0012. Return to text.
- “There is no evidence that effectively neutral mutations are heavily biased toward being very slightly deleterious.” discourse.peacefulscience.org/t/sft-on-genetic-entropy/11638/199. Return to text.
- “Instead, an actual genome is one randomly chosen configuration from among all of the selectively equivalent configurations, and effectively neutral mutations take move it to other selectively equivalent configurations. The precise fitness of the genome will drift up and down very slightly, but changes are as likely to be positive as negative.”discourse.peacefulscience.org/t/sft-on-genetic-entropy/11638/247. Return to text.
- “As for weakly selected mutations whose selection coefficients are less than 1/(4N) in absolute value, it is very very hard to collect information on what fraction of those are deleterious.” discourse.peacefulscience.org/t/paul-price-what-are-the-substantive-critiques-of-genetic-entropy/11736/23, in his point #5. Return to text.
- To be fair, Kimura didn’t really believe there were no beneficial mutations, he simply didn’t attempt to model them in relation to deleterious ones. He speculated that mega-beneficial mutations would negate entropic decay, but didn’t support that claim with any logic or evidence. Dr Sanford addressed mega-beneficial mutations in his book and subsequent publications. Return to text.
- “More or less, yes. Keep in mind that we know far more about the genome than Kimura did.” (In response to being asked if the pictured distribution is accurate). discourse.peacefulscience.org/t/the-distribution-of-the-effects-of-mutations/11869/2. Return to text.
- Sanford, J., Baumgardner, J., and Brewer, W., Selection threshold severely constrains capture of beneficial mutations; in: Marks II, R.J., Behe, M.J., Dembski, W.A., Gordon, B., and Sanford, J.C. (Eds.), Biological Information— New Perspectives, World Scientific, Singapore, pp. 264–297, 2013; worldscientific.com/doi/pdf/10.1142/9789814508728_0011. Return to text.
- “1. Yes, there is a disturbingly high mutation rate in humans, which would lead to a too-high mutational load. Possible explanations (a.) we’ve got the mutation rate wrong, or (b.) it was lower in prehistory, or (c.) the Earth is only 6,000 years old, and The End Is Nigh, and almost all of the geology and astronomy of the last 300 years is massively wrong. 2. It is not established that there is a comparable problem in all, or even most, other forms of life.” discourse.peacefulscience.org/t/paul-price-what-are-the-substantive-critiques-of-genetic-entropy/11736/23. Return to text.
- “You really do not seem to understand the situation here, or the essential reason that biologists (the ones who study real biology) don’t think there’s anything to Sanford’s claims. In a YEC world, all organisms start with perfect genomes. In that world, most mutations are indeed very slightly deleterious because (in your terms), we’re starting with perfectly designed machines and every change is for the worse. In an evolutionary world, in which all genomes are the result of mutation and selection, no genome was ever perfect. They’re not perfect machines. They’re Fords from the 1960s: poorly engineered, with lots of things that could be done better, but quite capable of functioning. In that world, most effectively neutral mutations aren’t damaging a perfect machine – they’re making insignificant differences to a machine that has always been imperfect. Every time you say something like ‘it’s much easier to damage a machine than improve it’ as though that settled the question, you demonstrate that you don’t understand what the question even is.” discourse.peacefulscience.org/t/sft-on-genetic-entropy/11638/417. Return to text.
- “8. The 2016 Current Opinions In Pediatrics article linked to by PDPrice is oblivious to the evidence for most of the genome being junk DNA.” discourse.peacefulscience.org/t/paul-price-what-are-the-substantive-critiques-of-genetic-entropy/11736/23 Return to text.
- The word gestalt means “leap” in German. We use it here as a hat-tip to former evolutionists, like Eldredge and Gould, who thought that evolution occurred in dramatic leaps. Return to text.
- Referring to Fig. 2 of Carter & Sanford’s peer-reviewed paper on H1N1. See Carter, R. and Sanford J.C., A new look at an old virus: patterns of mutation accumulation in the human H1N1 influenza virus since 1918, Theor. BiolMed. Model 9:42, 2012; tbiomed.biomedcentral.com/articles/10.1186/1742-4682-9-42 or More evidence for the reality of genetic entropy. Return to text.
- discourse.peacefulscience.org/t/paul-price-what-are-the-substantive-critiques-of-genetic-entropy/11736/14. Return to text.
- Pseudonym has been publicly acknowledged as belonging to Dr Stern Cardinale (e.g. at reddit.com/r/ChristianApologetics/comments/k3vskp/a_basic_introduction_to_genetic_entropy_at_long/ge7864h). Quote accurate as of 1 Dec 2020 (Note: Reddit users can modify comments at any time). reddit.com/r/ChristianApologetics/comments/k3vskp/a_basic_introduction_to_genetic_entropy_at_long/ge781a0/. Return to text.
- See, for example, Gibbs, A.J. et al. From where did the 2009 ‘swine-origin’influenza A virus (H1N1) emerge? Virology J 6:207, 2009. Return to text.
- Carter, R.W. More evidence for the reality of genetic entropy, J Creation 28(1):16–17, 2014; creation.com/evidence-for-genetic-entropy. Return to text.
- Carter, R.W. More evidence for the reality of genetic entropy—update, J Creation 33(1):3–4, 2019; creation.com/evidence-for-genetic-entropy-update. Return to text.



Readers’ comments
Comments are automatically closed 14 days after publication.